Resistin Induces Cardiomyocyte Hypertrophy in H9c2 Embryonic Rat Myocardial Cells via Inhibition of MiR-489-3p
-
Pengjie Yang
Department of Thoracic Surgery, The Affiliated People’s Hospital of Inner Mongolia Medical University, Hohhot, Inner Mongolia, Autonomous Region 010010, China
Jing HaoDepartment of Cardiology, Affiliated People’s Hospital of Inner Mongolia Medical University, Hohhot, Inner Mongolia, Autonomous Region 010010, China
Mingyu LiPanzhihua Key Laboratory of Cardiomyopathy and Heart Failure, Affiliated Hospital of Panzhihua University, Panzhihua, Sichuan 617099, China
Nengjun ZhaoPanzhihua Key Laboratory of Cardiomyopathy and Heart Failure, Affiliated Hospital of Panzhihua University, Panzhihua, Sichuan 617099, China
Lin Wu
Panzhihua Key Laboratory of Cardiomyopathy and Heart Failure, Affiliated Hospital of Panzhihua University, Panzhihua, Sichuan 617099, China
Background and Objective: The miRNAs play an important regulatory role in resistin-induced cardiac hypertrophy. The purpose of this experiment was to investigate the effect of resistin-induced myocardial hypertrophy in H9c2 cardiomyocytes by inhibiting the expression of miR-489-3p. Materials and Methods: After treatment with resistin or resistin plus miR-489-3p mimic, Image J was used to measure cell surface area. The BCA method and cell counting were used to measure protein synthesis. RT-qPCR was used to measure the relative expression levels of atrial natriuretic factor (ANF), brain natriuretic factor (BNF) and miR-489-3p. Results: Overexpression of the miR-489-3p reduced resistin-induced increase in cardiomyocyte area. Overexpression of miR-489-3p attenuated protein synthesis induced by resistin. Overexpression of miR-489-3p decreased the relative expression of cardiac hypertrophy marker ANF and BNF increased by resistin. Resistin inhibited the expression of miR-489-3p. Conclusion: Resistin induces cardiomyocyte hypertrophy by inhibiting the expression of miR-489-3p. This study provides a theoretical basis for further research on the role of miRNA in resistin-induced cardiomyocyte hypertrophy.
| Copyright © 2023 Yang et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
Heart failure (HF) is a chronic progressive clinical syndrome caused by abnormalities in the structure or function of the heart, resulting in impaired ventricular filling or ejection function, clinically manifested by dyspnea, fatigue and fluid retention, ultimately leading to cardiac exhaustion1,2. Cardiac hypertrophy, an early pathological change in heart failure, is a response to increased load-induced ventricular wall pressure to maintain cardiac output and persistent myocardial hypertrophy eventually leads to decompensation3,4. There are many reasons for myocardial hypertrophy, such as angiotensin II5, stress-induced6 or mediated by certain chemical substances7. Some adipokines are independent risk factors for cardiac hypertrophy8,9. Resistin is a cysteine-rich polypeptide, a member of adipokines with hormone-like activity and acts as an important signaling molecule in physiological and pathological processes such as obesity and metabolic syndrome10. Rodent resistin is mainly secreted by adipose tissue11. Different from rodents, human resistin is mainly from inflammatory cells12. Serum resistin is mainly secreted by macrophages and monocytes. In recent years, it has been suggested that resistin in humans may play an important role in the occurrence and development of cardiovascular events13,14. In a prospective study, intermediate-term mortality in patients undergoing coronary artery bypass therapy was found to be associated with resistin concentrations in perivascular adipose tissue15. Zhang et al.16 pointed out in a meta-analysis of the relationship between serum resistin concentration and hypertension that serum resistin levels in hypertensive patients were higher than those in normotensive controls, suggesting that resistin may be a risk factor for hypertension. Singh et al.17 reported that resistin induces cardiomyocytes to differentiate from fibroblasts into myofibroblasts via JNK/C-jun and JAK/STAT3 signaling pathways. Therefore, it is of great significance to clarify the signaling pathway of resistin-induced cardiomyocyte hypertrophy and its molecular mechanism for the prevention and improvement of cardiac hypertrophy.
The miRNAs are involved in the regulation of various pathophysiological processes of cardiomyocytes. They can affect the occurrence and development of cardiac diseases by regulating target proteins involved in the molecular pathways of excessive oxidative stress, apoptosis molecular pathways and endothelial cell dysfunction molecular pathways18,19. The miR-489-3p is a newly discovered small RNA closely related to cell proliferation, involved in muscle stem cell differentiation, invasion and migration of non-small cell lung cancer20 and also involved in the occurrence of various heart diseases, such as atherosclerosis, hypertrophic cardiomyopathy, heart failure and cardiac cachexia21. Recently, experimental studies indicate that miR-489-3p has an anti-cardiac hypertrophy effect in the study of aortic valve stenosis and hypertension-induced myocardial hypertrophy22. Yang et al.23 found that miRNA-489-3p could inhibit isoproterenol-induced cardiac fibrosis by downregulating histone deacetylase 2. However, whether miR-489-3p is involved in resistin-induced cardiomyocyte hypertrophy remains unclear. Therefore, the purpose of this study was to determine whether miR-489-3p was involved in resistin-induced cardiomyocyte hypertrophy.
MATERIALS AND METHODS
Reagents and solutions: This study was conducted from March 1, 2021 to March 1, 2022, in the Molecular Cancer Laboratory of People’s Hospital Affiliated with Inner Mongolia Medical University. The rat H9c2 cell line was purchased from China Concord Cell Bank (Beijing, China). Human Resistin was purchased from Peprotech Company (Rocky Hill, New Jersey and United States of America). The miR-489-3p Mimic was provided by Ruibo Company (Guangzhou, Guangdong and China). The ANF, BNF and miR-489-3p primers were designed and synthesized by Shanghai Sangon (Shanghai, China). The RNA extraction kits and reverse transcription kits were provided by Quanshijin Company (Beijing, China).
Cell culture and grouping: The H9c2 cardiomyocytes were cultured in a high-glucose complete medium containing 10% FBS. The medium was replaced every two to three days. When cells grew to 80-90%, they were divided into four groups. Con group (Control group), Res group (Resistin group), Mimic+Res group (MiR-489-3p mimic+resistin group), Mimic-NC+Res group (Mimic-negative control+resistin group). The 24 hrs after plating, cells were transfected with miR-489-3p mimic for 24 hrs and then replaced with a starvation medium containing resistin 50 ng mL–1 for 36 hrs.
Determination of cell surface area: After cells were treated with the corresponding drugs for a predetermined time, the cell morphology was observed under an inverted microscope and 3 different fields of view were randomly selected to take pictures. The 15 cells were randomly selected in each field of view and cell surface areas were determined with ImageJ software (1.49 version, NIH, Bethesda, Maryland, United States of America)24.
Determination of protein synthesis: Cells in each group were treated with different drugs for a predetermined time and then trypsinized for 3 min. Cells were counted under a microscope (I×51, Olympus, Tokyo, Japan). The 100 μL of RIPA (Strong RIPA: PMSF = 100: 1) were added to cells. Cells were lysed on ice and centrifuged with a high-speed refrigerated centrifuge (DH-20KR, Gallop Technology, Shanghai, China). The supernatant was aspirated to be measured. The protein concentration was determined by the BCA method24. A standard curve was drawn according to the known standard protein concentration and absorbance value. Protein synthesis is expressed single cell protein content.
Transfection of miR-489-3p mimic: MiR-489-3p mimic and Mimic-NC dry powder were briefly centrifuged and dissolved in 250 μL of Rnase-free H2O or sterilized ddH2O to prepare 20 μM of stock solutions, respectively, which were stored at -80°C refrigerator in aliquot to avoid repeated freezing and thawing (no more than five times). Cells were seeded in six-well plates for 24 hrs. When transfecting cells, 5 μL of 20 μM miR-489-3p mimic or Mimic-NC with 120 μL of riboFECT™ CP Buffer was diluted and mixed gently. The 12 μL of riboFECT™ CP Reagent was added and incubated at room temperature for 15 min. The above mixture was added into 1863 μL of DMEM medium and mixed gently to make the final concentration of miR-489-3p mimic or Mimic-NC 50 nM. After transfection, cells were incubated at 37°C for 24 hrs. The sequence of miR-489-3p mimic and negative control were as follows:
• |
miR-489-3p mimic forward, 5'-AUGACAUCACAUAUAUGGCAGC-3' and reverse, 5'-GCUGCCAUAUAUGUGAUGUCAU-3' |
|
• |
Mimic-NC forward, 5'-UUUGUACUACACAAAAGUACUG-3' and reverse, 5'-CAGUACUUUUGUGUAGUACAAA-3' |
RT-qPCR: Total cellular RNA was extracted with the TransZol Up kit. TransScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR kit (For ANF, BNF reverse transcription) and TransScript miRNA First-Strand cDNA Synthesis SuperMix kit (For miR-489-3p reverse transcription) were used for reverse transcription. PerfectStarTM Green qPCR SuperMix kit was used for amplification. The preparation of the reaction system was carried out according to the instructions in the kit (Germany). In the PCR instrument (Analytik jena AG, Jena, Germany), firstly, 94°C for the 30 sec, then 94°C for 5 sec, cooling to 60°C for 30 sec and repeated cycles for 40 times and the fluorescence was captured at the end of 60°C for 30 sec. RT-qPCR reaction was performed using the ABI OneStep system (Thermo Fisher Scientific Inc. Waltham, Morocco, United States of America). The expression of miR-489-3p took U6 as the internal reference. The expressions of ANF and BNF took GAPDH as the internal reference. The relative expression levels were calculated using 2–ΔΔCT. The kits used were purchased from Beijing Quanzhijin Co., Ltd. (Beijing, China).
The primers were designed as follows:
BNF-F: 5'-ACAATCCACGATGCAGAAGC-3'
BNF-R: 5'-TAGGGCCTTGGTCCTTTGAG-3'
ANF-F: 5'-CGGACAAAGGCTGAGAGAGA-3'
ANF-R: 5'-ACCGCACTGTATACGGGATT-3'
miR-489-3p-F: 5'-CATGACATCACATATATGGCAGC-3'
miR-489-3p-R: 5'-ATCCAGTGCAGGGTCCGAGG-3'
Statistical analysis: All experimental data were analyzed and processed using SPSS 21.0 professional statistical software and expressed as Mean±Standard deviation. One-way ANOVA was used to compare
data between multiple groups. Data were compared between two groups using the t-test. The p<0.05 indicated that the difference was statistically significant. Article graphs were drawn with GraphPad Prism 8.0.
Ethical consideration: This study was conducted using the H9c2 cell line purchased from the company. This study didn’t involve human rights, animal rights or other related ethics.
RESULTS
Expression of miR-489-3p in resistin-induced cardiomyocyte hypertrophy: Cells were treated with resistin for 36 hrs. RNA was extracted and reverse transcribed to cDNA for real-time quantitative PCR to detect the relative expression of miR-489-3p in each experimental group (U6 was used as an internal reference). Compared with the Con group, the expression of miR-489-3p in the Res group was significantly decreased (0.5037±0.07961 vs. 1, p<0.01). Mimic+resistin could increase the expression of miR-489-3p compared with the Res group (0.7020±0.07858 vs. 0.5037±0.07961 and p<0.05). Whereas, Mimic-NC+resistin had no difference compared with the Res group (Fig. 1).
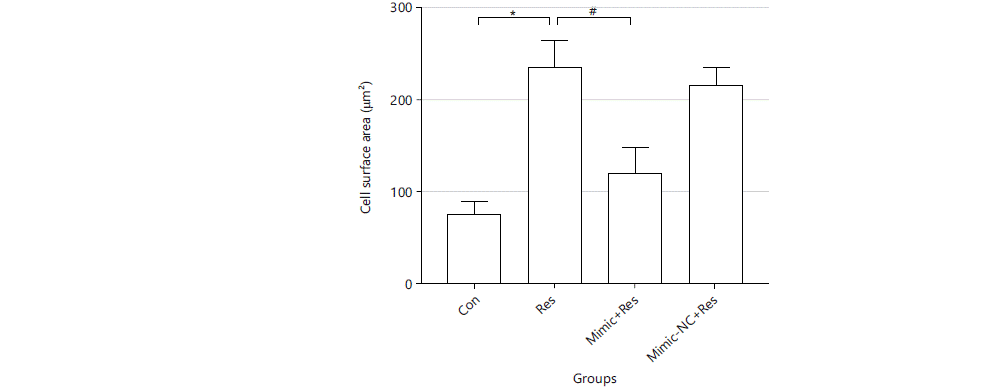
Regulatory effect of miR-489-3p in resistin-induced changes in cardiomyocyte area: After cells were treated accordingly, the adherent growth of cells was observed, with a long spindle shape, abundant cytoplasm and differences in the cell surface area. Cell surface areas were as follows. Con group: 77.153±11.341, Res: 236.714±27.861, Mimic+Res group: 120.908±26.144, Mimic-NC+Res group: 217.623±19.197, unit: μm2. Compared with the Con group, the cell surface area of the Res group was significantly increased (p<0.01). Compared with the Res group, the cell surface area of the Mimic+Res group was significantly lower than that of the Res group (p<0.01). There were no significant differences in cell surface area between the Res group and the Mimic-NC+Res group (Fig. 2).
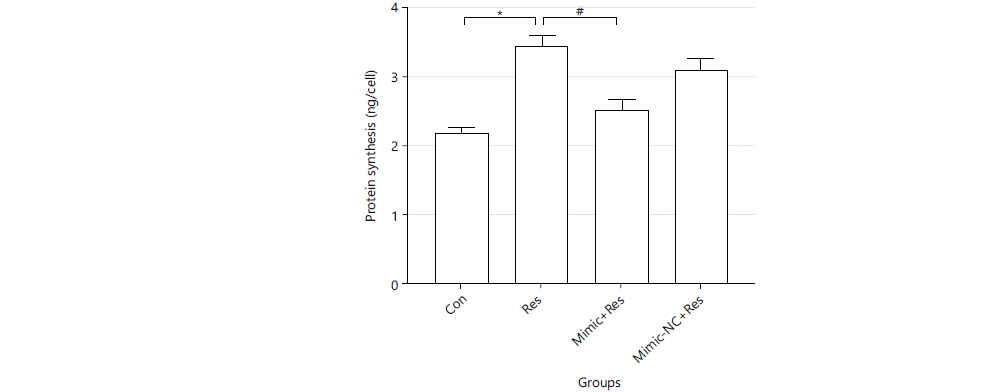
Regulatory effect of miR-489-3p in resistin-induced protein synthesis: Resistin could induce increased protein synthesis in cardiomyocytes, which could be improved by overexpression of miR-489-3p. Cells were treated accordingly for 36 h, protein synthesis was measured. The results were as follows: Con group: 2.221±0.056, Res: 3.455±0.171, Mimic+Res group: 2.544±0.143, Minic-NC+Res group: 3.123±0.167 unit: ng/cell. Compared with the Con group, the cellular protein synthesis of the Res group was significantly increased (p<0.01). The cellular protein synthesis of the Mimic+Res group was significantly lower than that of the Res group (p<0.01) (Fig. 3).
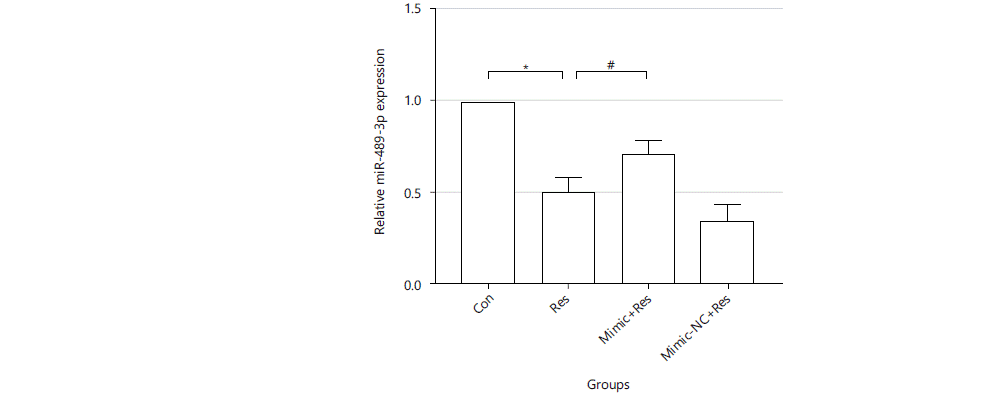
|
|
|
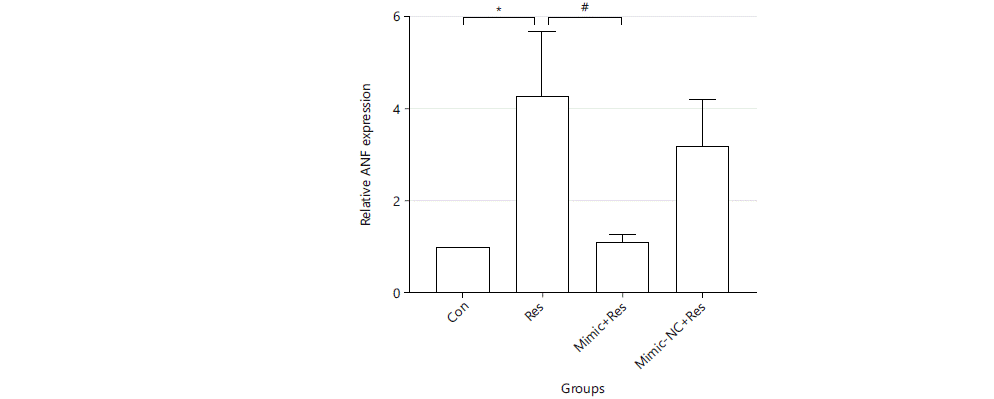
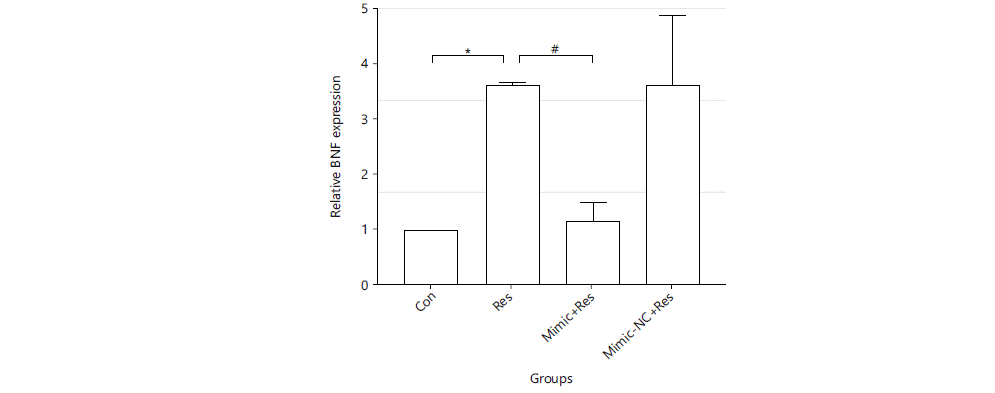
Regulatory effect of miR-489-3p in resistin-induced cardiomyocyte hypertrophy genes: Resistin could induce increased expression of cardiomyocyte hypertrophy genes in cardiomyocytes, which could be decreased by overexpression of miR-489-3p. After the cells were treated for 36 hrs, RNA was extracted and reverse transcribed and real-time quantitative PCR was performed to detect the relative expression levels of cardiac hypertrophy marker gene ANF and BNF (GAPDH was used as an internal reference). Compared with the Con group, the expression of cardiac hypertrophy marker genes in the Res group was significantly increased (ANF: 4.326±1.433 vs. 1, BNF: 3.661±0.07261 vs. 1. p<0.01). The expression of ANF and BNF in the Mimic+Res group was significantly decreased compared with the Res group (ANF: 1.149±0.1371 vs. 4.326±1.433, BNF: 1.171±0.3372 vs. 3.661±0.07261 and p<0.01) (Fig. 4 and 5).
|
|
DISCUSSION
In this study, it was found that 50 ng mL–1 resistin could induce cardiomyocyte hypertrophy after 36 hrs. It increased myocardial cell surface area, protein synthesis and cardiac hypertrophy marker ANF and BNF expression. miR-489-3p was involved in resistin-induced cardiomyocyte hypertrophy. These findings are consistent with those of most researchers. Resistin is involved in many life processes such as cellular energy metabolism, proliferation and autophagy. Resistin is not only expressed in adipose tissue, liver and tumor tissue but also exists in blood circulation in a free form. Resistin is involved in the pathogenesis of cardiovascular disease. Plasma resistin levels are closely related to the development of atherosclerosis, acute myocardial infarction and heart failure25. Resistin is an independent predictor of adverse cardiovascular events26. Some experiments exhibit that resistin can promote cardiomyocyte hypertrophy27,28. Previous studies have found that resistin can induce cardiomyocyte hypertrophy, but its molecular mechanism is still not fully revealed. Liu et al.29 found that resistin-induced H9c2 cardiomyocyte hypertrophy by inhibiting the LKB1/AMPK signaling pathway. Metformin could stimulate the LKB1/AMPK signalling pathway to improve cardiomyocyte hypertrophy. Luo et al.30 reported that apelin could reverse resistin-induced cardiomyocyte hypertrophy by inhibiting the expression of ERK1/2.
In this study, it was found that resistin inhibited the expression of miR-489-3p. Recent studies have concluded that many miRNAs are involved in the regulation of cardiac hypertrophy31-33 including miR-2234, miR-133a35 and miR-208a36. Ucar et al.37 found that mice lacking miR-212/132 were protected from pressure overload-induced heart failure. Overexpression of miR-212/132 resulted in pathological cardiac hypertrophy, heart failure and even death in mice. Shi et al.38 reported that the expression of miR-26a-5p was down-regulated in myocardial hypertrophy induced by thoracic aortic coarctation or in angiotensin II-induced H9c2 cardiomyocyte hypertrophy. Overexpression of miR-26a-5p could alleviate cardiac hypertrophy by targeting ADAM17. Wang et al.22 found that expression of miR-489-3p was decreased in angiotensin II-induced cardiomyocyte hypertrophy. miR-489-3p downstream signaling protein was MyD88. Increasing the expression of miR-489-3p or inhibiting MyD88 expression could reverse cardiomyocyte hypertrophy. This was consistent with current findings. In this study, it was found that the expression of miR-489-3p was significantly lower in the resistin group than in the control group. Myocardial cell surface area, protein synthesis and cardiac hypertrophy gene expression were significantly increased in the resistin group, suggesting that resistin-induced cardiomyocyte hypertrophy. In contrast, the miR-489-3p mimic+Res group obviously increased the expression of miR-489-3p compared with the resistin group. MiR-489-3p mimic+Res group significantly decreased cell surface area, protein synthesis and cardiomyocyte embryonic gene expression. It was suggested that increasing the expression of miR-489-3p can reverse resistin-induced myocardial hypertrophy.
In this study, the relationship between resistin-induced cardiac hypertrophy and miR-489-3p was investigated. Resistin-induced cardiomyocyte hypertrophy via inhibiting the expression of miR-489-3p, which clarified that miR-489-3p may serve as a target for regulating cardiac hypertrophy, laying the foundation for the prevention and treatment of resistin-induced cardiac hypertrophy in the clinic. The limitation of this study is that the targets of miR-489-3p were not further investigated. In addition, it was only studied at the cellular level, lacking animal experiments to validate it from the in vivo level and more in-depth studies are still needed.
CONCLUSION
In conclusion, resistin inhibited miR-489-3p expression, whereas miR-489-3p suppressed resistin-induced cardiomyocyte hypertrophy. It was shown that resistin induced cardiomyocyte hypertrophy by suppressing miR-489-3p expression. This study helps to further investigate the role of miRNAs in resistin-induced cardiomyocyte hypertrophy and provides new targets and methods for the clinical application of miRNAs in the treatment of resistin-induced cardiomyocyte hypertrophy.
SIGNIFICANCE STATEMENT
In this study, the results showed that resistin induced cardiomyocyte hypertrophy by suppressing the expression of miR-489-3p. It helps researchers to further investigate the role of other miRNAs in resistin-induced cardiac hypertrophy. This study may partially elucidate the molecular mechanism of resistin-induced cardiac hypertrophy and provide new ideas and targets for the treatment of cardiac hypertrophy and heart failure.
ACKNOWLEDGMENTS
Pengjie Yang and Jing Hao contributed to this study equally. This study was supported by Inner Mongolia Natural Science Foundation, China (Grant No. 2020MS08107 and 2020MS08187), Inner Mongolia Autonomous Region Science and Technology Program, China (Grant No. 2019GG107), High-end Talent Introduction Project of Sichuan Provincial Department of Science and Technology, China (Grant No. 2022JDGD0034), Medical Science and Technology Project of Sichuan Provincial Health Commission, China (Grant No. 21PJ200), Provincial-level science and Technology Program Transfer Payment Special Fund project of Panzhihua Science and Technology Bureau, grant number 222ZYZF-S-01.
REFERENCES
- Greene, S.J., G.C. Fonarow and J. Butler, 2020. Risk profiles in heart failure baseline, residual, worsening, and advanced heart failure risk. Circ: Heart Fail., 13.
- Snipelisky, D., S.P. Chaudhry and G.C. Stewart, 2019. The many faces of heart failure. Cardiac Electrophysiol. Clin., 11: 11-20.
- Nakamura, M. and J. Sadoshima, 2018. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol., 15: 387-407.
- Oldfield, C.J., T.A. Duhamel and N.S. Dhalla, 2020. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can. J. Physiol. Pharmacol., 98: 74-84.
- Shen, Y., X. Wang, R. Yuan, X. Pan and X. Yang et al., 2021. Prostaglandin E1 attenuates AngII-induced cardiac hypertrophy via EP3 receptor activation and Netrin-1upregulation. J. Mol. Cell. Cardiol., 159: 91-104.
- Zeng, Y., W.Q. Ren, A.Z. Wen, W. Zhang, F.Y. Fan and O.Y. Chen, 2022. Autophagy and pressure overload-induced cardiac hypertrophy. J. Asian Nat. Prod. Res., 24: 1101-1108.
- Kang, S., E.R. Chemaly, R.J. Hajjar and D. Lebeche, 2011. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J. Biol. Chem., 286: 18465-18473.
- Fontes-Carvalho, R., M. Fontes-Oliveira, F. Sampaio, J. Mancio and N. Bettencourt et al., 2014. Influence of epicardial and visceral fat on left ventricular diastolic and systolic functions in patients after myocardial infarction. Am. J. Cardiol., 114: 1663-1669.
- Kankaanpää, M., H.R. Lehto, J.P. Pärkkä, M. Komu and A. Viljanen et al., 2006. Myocardial triglyceride content and epicardial fat mass in human obesity: Relationship to left ventricular function and serum free fatty acid levels. J. Clin. Endocrinol. Metab., 91: 4689-4695.
- Tripathi, D., S. Kant, S. Pandey and N.Z. Ehtesham, 2020. Resistin in metabolism, inflammation, and disease. FEBS J., 287: 3141-3149.
- Huang, X. and Z. Yang, 2016. Resistin’s, obesity and insulin resistance: The continuing disconnect between rodents and humans. J. Endocrinol. Invest., 39: 607-615.
- Li, Y., Q. Yang, D. Cai, H. Guo and J. Fang et al., 2021. Resistin, a novel host defense peptide of innate immunity. Front. Immunol., 12.
- Rachwalik, M., M. Hurkacz, B. Sienkiewicz-Oleszkiewicz and M. Jasiński, 2021. Role of resistin in cardiovascular diseases: Implications for prevention and treatment. Adv. Clin. Exp. Med., 30: 865-874
- Recinella, L., G. Orlando, C. Ferrante, A. Chiavaroli, L. Brunetti and S. Leone, 2020. Adipokines: New potential therapeutic target for obesity and metabolic, rheumatic, and cardiovascular diseases. Front. Physiol., 11.
- Tonhajzerova, I., I. Ondrejka, N. Ferencova, I. Bujnakova and M. Grendar et al., 2021. Resistin levels in perivascular adipose tissue and mid-term mortality in patients undergoing coronary artery bypass granting. Physiol. Res., 70: 543-550.
- Zhang, Y., Y. Li, L. Yu and L. Zhou, 2017. Association between serum resistin concentration and hypertension: A systematic review and meta-analysis. Oncotarget, 8: 41529-41537.
- Singh, R., R.K. Kaundal, B. Zhao, R. Bouchareb and D. Lebeche, 2021. Resistin induces cardiac fibroblast-myofibroblast differentiation through JAK/STAT3 and JNK/c-Jun signaling. Pharmacol. Res., 167.
- Çakmak, H.A. and M. Demir, 2020. MicroRNA and cardiovascular diseases. Balkan Med. J., 37: 60-71.
- Zhou, S.S., J.P. Jin, J.Q. Wang, Z.G. Zhang, J.H. Freedman, Y. Zheng and L. Cai, 2018. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin., 39: 1073-1084.
- Dao, R., M. Wudu, L. Hui, J. Jiang, Y. Xu, H. Ren and X. Qiu, 2020. Knockdown of lncRNA MIR503HG suppresses proliferation and promotes apoptosis of non-small cell lung cancer cells by regulating miR-489-3p and miR-625-5p. Pathol. Res. Pract., 216.
- Wojciechowska, A., A. Osiak and K. Kozar-Kamińska, 2017. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med., 26: 868-874.
- Wang, K., F. Liu, L.Y. Zhou, B. Long and S.M. Yuan et al., 2014. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res., 114: 1377-1388.
- Yang, X., T. Yu and S. Zhang, 2020. MicroRNA‑489 suppresses isoproterenol‑induced cardiac fibrosis by downregulating histone deacetylase 2. Exp. Ther. Med., 19: 2229-2235.
- Luo, J., H. Liu, X. Zheng, B. Lin and Q. Ye et al., 2019. Inhibitory effect of apelin on cardiomyocyte hypertrophy induced by resistin in H9c2 cells. Int. J. Pharmacol., 15: 311-317.
- Abate, N., H. Sallam, M. Rizzo, D. Nikolic, M. Obradovic, P. Bjelogrlic and E. Isenovic, 2014. Resistin: An inflammatory cytokine. Role in cardiovascular diseases, diabetes and the metabolic syndrome. Curr. Pharm. Des., 20: 4961-4969.
- Fontana, A., S. Spadaro, M. Copetti, B. Spoto and L. Salvemini et al., 2015. Association between resistin levels and all-cause and cardiovascular mortality: A new study and a systematic review and meta-analysis. PLoS ONE, 10.
- Kim, M., J.K. Oh, S. Sakata, I. Liang, W. Park, R.J. Hajjar and D. Lebeche, 2008. Role of resistin in cardiac contractility and hypertrophy. J. Mol. Cell. Cardiol., 45: 270-280.
- Hussain, S., M. Asghar and Q. Javed, 2010. Resistin gene promoter region polymorphism and the risk of hypertrophic cardiomyopathy in patients. Transl. Res., 155: 142-147.
- Liu, P., G.C. Cheng, Q.H. Ye, Y.Z. Deng and L. Wu, 2016. LKB1/AMPK pathway mediates resistin-induced cardiomyocyte hypertrophy in H9c2 embryonic rat cardiomyocytes. Biomed. Rep., 4: 387-391.
- Luo, J.W., X. Zheng, G.C. Cheng, Q.H. Ye, Y.Z. Deng and L. Wu, 2016. Resistin-induced cardiomyocyte hypertrophy is inhibited by apelin through the inactivation of extracellular signal-regulated kinase signaling pathway in H9c2 embryonic rat cardiomyocytes. Biomed. Rep., 5: 473-478.
- Wang, J. and X. Yang, 2012. The function of miRNA in cardiac hypertrophy. Cell. Mol. Life Sci., 69: 3561-3570.
- Nie, X., J. Fan, H. Li, Z. Yin and Y. Zhao et al., 2018. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol. Ther. Nucleic Acids, 12: 254-266.
- Wang, J., O. Liew, A. Richards and Y.T. Chen, 2016. Overview of microRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int. J. Mol. Sci., 17.
- Tu, Y., L. Wan, L. Bu and B. Shen, 2017. Retraction of: Abstract P71, in vitro and in vivo direct monitoring of miRNA-22 expression in isoproterenol-induced cardiac hypertrophy by bioluminescence imaging. Cardiovasc. Res., 113: 248-248
- Xiao, Y., J. Zhao, J.P. Tuazon, C.V. Borlongan and G. Yu, 2019. MicroRNA-133a and myocardial infarction. Cell Transplant., 28: 831-838.
- Huang, X.H., J.L. Li, X.Y. Li, S.X. Wang and Z.H. Jiao et al., 2021. miR-208a in cardiac hypertrophy and remodeling. Front. Cardiovasc. Med., 8: 773314.
- Ucar, A., S.K. Gupta, J. Fiedler, E. Erikci and M. Kardasinski et al., 2012. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun., 3.
- Shi, H., H. Li, F. Zhang, H. Xue, Y. Zhang and Q. Han, 2021. MiR-26a-5p alleviates cardiac hypertrophy and dysfunction via targeting ADAM17. Cell Biol. Int., 45: 2357-2367
How to Cite this paper?
APA-7 Style
Yang,
P., Hao,
J., Li,
M., Zhao,
N., Wu,
L. (2023). Resistin Induces Cardiomyocyte Hypertrophy in H9c2 Embryonic Rat Myocardial Cells via Inhibition of MiR-489-3p. Trends in Medical Research, 18(1), 92-101. https://doi.org/10.3923/tmr.2023.92.101
ACS Style
Yang,
P.; Hao,
J.; Li,
M.; Zhao,
N.; Wu,
L. Resistin Induces Cardiomyocyte Hypertrophy in H9c2 Embryonic Rat Myocardial Cells via Inhibition of MiR-489-3p. Trends Med. Res 2023, 18, 92-101. https://doi.org/10.3923/tmr.2023.92.101
AMA Style
Yang
P, Hao
J, Li
M, Zhao
N, Wu
L. Resistin Induces Cardiomyocyte Hypertrophy in H9c2 Embryonic Rat Myocardial Cells via Inhibition of MiR-489-3p. Trends in Medical Research. 2023; 18(1): 92-101. https://doi.org/10.3923/tmr.2023.92.101
Chicago/Turabian Style
Yang, Pengjie, Jing Hao, Mingyu Li, Nengjun Zhao, and Lin Wu.
2023. "Resistin Induces Cardiomyocyte Hypertrophy in H9c2 Embryonic Rat Myocardial Cells via Inhibition of MiR-489-3p" Trends in Medical Research 18, no. 1: 92-101. https://doi.org/10.3923/tmr.2023.92.101

This work is licensed under a Creative Commons Attribution 4.0 International License.