Principles, Applications and Limitations of the Liquisolid System of Drug Delivery: A Review
-
Daniel Ekpa Effiong


Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, University of Uyo, Nigeria
Godswill Chukwunweike OnunkwoDepartment of Pharmaceutical Technology and Industrial Pharmacy, Faculty of Pharmaceutical Sciences, University of Nigeria, Nsukka, Nigeria
| Received 19 Apr, 2024 |
Accepted 07 Jun, 2024 |
Published 08 Jun, 2024 |
The liquisolid system of drug delivery continues to find usefulness as a potent means to circumvent solubility and dissolution challenges commonly encountered during drug formulation and medicine development in the pharmaceutical industry. Originally invented by Spireas, the technique has gained acceptance and improved over the years, in the types of excipients used, nature and class of drug formulated and its applications not only in transforming lipophilic and poorly soluble drugs to solid dosage forms with enhanced bioavailability, but also now gaining popularity in modified delivery, photoprotection and minimizing the effect of pH on drug release. This review thus highlights the foundational principles of the liquisolid technique, examines current methods employed and modern applications of this technique. It also gives present knowledge of this delivery technique and its limitations. A systematic approach to the search of published literature in standard databases (Pubmed, Google Scholar, Researchgate) was carried out using specific search terms and operators that provided available works as information sources for this review. Critical evaluation of obtained literature from the database was adopted to extract data on the principles and current applications of the liquisolid systems beyond its intended initial use. The prevailing mechanism for enhancing bioavailability via the technique is improved wetting and drug presentation in well dispersed or soluble state; minimal pH influence is based on the phenomenon of saturation solubility of the drug in the nonvolatile vehicle; and photoprotection is due to high refractive and diffraction capacities of the coating materials used. This work concludes by presenting the identified and potential limitations of the liquisolid technique. It gives expert opinions on how these are being circumvented and future perspectives on this ever-unfolding promising drug delivery strategy. It will be an invaluable contribution to current literature in drug development research employing liquisolid techniques for drug delivery.
| Copyright © 2024 Effiong and Onunkwo. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
The growing interest in the use of liquisolid systems in drug formulation and delivery studies has been related to its invaluable contribution to circumventing poor drug solubility, enhancing bioavailability and presenting several delivery strategies with desirable therapeutic outcomes1.
Solubility, dissolution and bioavailability rank highly as parameters considered during the development of potential therapeutic agents for use as medicines because sooner or later, all medicines must be in solution to be absorbed or reach desired drug concentration in systemic circulation for therapeutic efficacy2,3. However, the successful transformation of a potent drug into medicine is often beset by challenges of these physicochemical parameters or those related to them. Solubility and dissolution rate of a drug can thus significantly influence its absorption, determine its bioavailability in the circulatory system and expectedly influence the expected pharmacological response1,4.
Drugs with poor water solubility are generally formulated and used as high-dose medicines with high dosage regimens so that, after administration (for example via oral route), the fraction of it that goes into solution could reach therapeutic plasma concentrations5. Sadly, high-dose medicines could mean exceeding the safety limits of the drugs and approaching toxicity, escalated untoward effects and increased production costs. As medicinal chemists and formulation scientists continually search for and employ strategies to improve the rate and amount of poorly soluble drugs that go into solution, the liquisolid system of drug delivery is now gaining attention. Thus in this paper, this technique will be well explored in terms of its principles, its application and limitations associated with it. This work will also address how these limitations are being overcome. First, a background of poor drug solubility will be presented, how widespread the problem is and common strategies that have been employed to improve it will be highlighted.
Generally, when discussing solubility and dissolution studies of drugs, the solvent of interest is usually an aqueous medium or one that is water-miscible because it is the main component of human body fluids where administered drugs will dissolve and subsequently be absorbed. This also explains why it is also the widely used solvent for liquid pharmaceutical formulations. In medicine development and manufacture of new chemical entities and candidates, the unprecedented incidence of low aqueous solubility is a major problem. One possible reason is that most drugs developed as medicines are either weakly acidic or weakly basic, thus having poor aqueous solubility. Yet these drugs must be present in an aqueous solution at the site of absorption to be taken up by the body and utilized.
The recent unlimited leap in the sphere of drug discovery resulting from the interplay of combinatorial chemistry, high throughput screening and computer-aided-drug designs and modeling has made possible rapid drug synthesis, optimization and drug-receptor simulation studies in silico6. This progress has churned out a number of drug molecules that can effectively and selectively interact with receptors (or ligands) within the body to elicit biological activity. These resulting available drug molecules or candidates, however, are highly hydrophobic, show a slow dissolution rate and are poorly water soluble2,7. No doubt, an indication of the magnitude of solubility challenges in the development and manufacture of medicines.
CHALLENGE OF POOR SOLUBILITY- HOW EXTENSIVE!
On the basis of their dissolution, water solubility and permeability in the intestinal tract, medicines are being classified by the Biopharmaceutical Classification System. Using this system and well-defined parameters, medicines are classified into Class I (well absorbed; that is have high solubility and permeability) Class II (limited solubility, high permeability low solubility) Class III (limited permeability, having high solubility but low permeability) and Class IV (poorly absorbed, having both solubility and permeability as being low)8. The parameters as defined of the dose of the medicine as follows; solubility (in 250 mL or less of water over a pH range of 1-8), dissolution rate (in dissolution apparatus 1 at 100 rpm or apparatus 2 at 50 rpm, in a volume of 900 mL buffer solutions of 0.1 N HCl/pH 4.5 buffer/pH 6.8 buffer with no enzymes) and permeability (using absorption rate of the prescribed dosage and is stability in the stomach).
Reportedly, some 4 out of every 10 medicines already in the market are poorly water soluble and about 90% of newly approved drug candidates in development are poorly water soluble. They fall in Class II and IV of the Biopharmaceutical Classification System and many of these fail to progress in the line to realization as medicines it is on record that almost 70-80% of the synthesized chemical entities have been classified as having poor solubility2,3,7. These statistics are clear indications of how extensive the issue of poorly water-soluble drugs and this is understandably a matter of serious concern to the formulation scientist, who constantly attempts to explore formulation strategies to circumvent this challenge but positively influence drugs to achieve medicines with enhanced delivery.
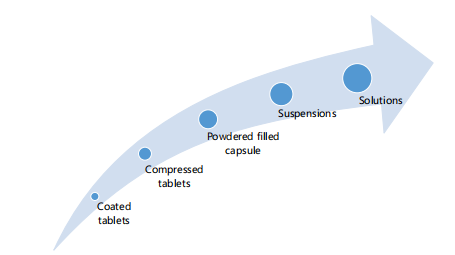
Solubility is not to be interchanged as dissolution or dissolution rate. Dissolution is the amount of a substance that goes into a solution at a time. Dissolution rate is described as the amount of a substance (drug) that goes into solution over a given time but solubility defines the total amount of the same drug that can go into solution under specified conditions2. Solubility is not a function of time. As a general rule, poor aqueous soluble substances have a slow dissolution rate, especially in coarse dispersions but an exception is hydroxypropyl methylcellulose, for instance, though highly soluble excipient for drug formulation has a very slow rate of dissolution because it takes a relatively longer time to be fully wetted to go into solution9. In the same vein, different dosage forms or delivery systems go into solution to exert therapeutic action at varying rates depending on how they were designed to function. Figure 1 presents different dosage forms and their rate of dissolution in increasing order in the direction of the arrow.
Strategies for improving solubility and dissolution- the associated limitations: For poorly soluble drugs or hydrophobic ones, several conventional methods and new formulation techniques have been employed by researchers and medicine manufacturers to overcome poor solubility and dissolution. These techniques and strategies as found in the published literature on improving solubility are numerous and can be grouped into physical techniques (by modification of particle sizes to micro sizes and nano-based systems thus increasing particle-specific surface area for improved dissolution rate); chemical modifications (use of different salt form, pH adjustment) and other formulation techniques. Table 1 presents a summary of such approaches, their application in particular drugs and their respective limitations.
Challenges associated with other techniques of improving poor solubility: As reflected in the table, these strategies to improve solubility and dissolution of drugs having challenging solubility profiles come with their limitations. Beyond those listed, some methods are even capital intensive, require sophisticated equipment, or demand technology. This is especially so in methods such as soft gelatin encapsulation, where poor soluble drugs are prepared in aqueous or oily solution form and then encapsulated as advanced solid dosage forms.
|
| Table 1: | Summary of the techniques for enhanced solubility and their limitations | |||
| Solubility enhancing techniques |
Process involved | Application | Limitation | Reference |
| Chemical modifications | ||||
| Salt formation | Conversion of drug to a salt form that is water soluble |
Diclofenac manufactured as salts of sodium/potassium |
Formation of new product with likely new properties and |
Patil et al.10 |
| Amlodipine as besylate salt Theophylline, Barbiturates |
requiring safety assessment |
Samineni et al.8 | ||
| pH adjustment | Used to enhance solubility of ionizable organic medicines in solutions Use of buffers |
Aspirin tablets are used as dissolving tablets by preparing with sodium bicarbonate and citric acid as pH adjusting buffers |
Limited application to drugs that are weakly acidic or basic |
Kalepu and Nekkanti11 |
| In situ salt formation | Ciprofloxacin infusions formed by adjusting pH with lactic acid |
|||
| Prodrug | Approach converts a pharmacologically active substance into an inactive one that gets activated in the system after absorption through enzyme or chemical action |
Paclitaxel delivered as Paclitaxel-disulfide prodrug, for improved anticancer activity Chalcone delivered as Chalcone-phosphate |
Requires certain specific conditions for the active drug the active drug to be released from the prodrug e.g., presence of an hydrolytic enzyme to release the drug after administration New moieties may come with newer physicochemical properties |
Jornada et al.12 |
| Physical methods | ||||
| Solid dispersion | Hydrophobic drugs are dispersed in solid excipients matrix which is hydrophilic |
Delivery of Paclitaxel an anticancer, delivery of curcumin and curcumin complex for acetylcholine esterase inhibition and anticancer, respectively |
Limited commercial application because of poor stability on storage and limited understanding of product solid-state structure |
Wu et al.13, de Sá et al.14 and Choi et al.15 |
| Encapsulation of liquid drug using soft gelatin |
Drug in liquid form is coated or protected by capsule or covering It has the highest and most consistent bioavailability, mainly due to the fact that the drug is already in solution |
Lipid soluble vitamins | Expensive and uncommon technology |
Arango-Ruiz et al.16 |
| Self nanoemulsion (SNEDDS), self micro- emulsifying drug (SMEDDS) or self- emulsifying drug delivery system (SEDDS) |
Drug is prepared to be carried within rapidly forming emulsification globules formed when in contact with the fluid in the gastrointestinal tract The size of the globules is used to classify the delivery system as SEDDs (if macro) SMEDDS (if micron sized or SNEDDS if nanosized |
Atorvastatin self- globules of emulsion had better Cyclosporine an immunosuppressant the management of benign prostatic hyperplasia |
High cost and complicated procedure |
Shen and Zhong17 Kalepu and Nekkanti11 and Agubata et al.18 |
| Solid lipid nanoparticles | Drugs are held within the lipid matrices Site-specific delivery achievable |
Ofloxacin was formulated as SLN fenofibrate successful incorporation into SLN |
High cost, a process complicated |
Xie et al.19 |
| Microencapsulation | A packaging engineering te unique in that drugs are coated with thing outer polymer layer as shells to form microcapsules |
Delivery of doxorubicin and heparin using chitosan for encapsulation in managing human papilloma carcinoma |
Selecting suitable coating polymer requires good technical skill |
Chaturvedi and Sharma20 |
| Size reduction | ||||
| Micronization and nanonization |
Micronization: Drug is sized reduced by milling to micrometer size ranges precipitation to obtain submicron size ranges so as to increase the surface area and enhance solubility |
Micronization is employed in the delivery of ibuprofen to improve digestive absorption and bioavailability |
Micronization: High |
Han et al.21 |
| Nanosuspension | Nanosuspension: |
Nanosuspension: Delivery of megesterol acetate (Megace ES®) for rapid dissolution and bioavailability Delivery of fenofibrate tablet (Tricor®) for improved (Tricor®) for improved dissolution and bioavailability |
Nanosuspension: Formulation design and stabilization is complex No controlled release |
Kanikkannan6 |
| Use of technological advancement | ||||
| Inclusion complexation | Involves inserting a nonpolar molecule (or poorly soluble drug) into the cavity of another, the host/companion molecule (eg cyclodextrin) and held by non-covalent intermolecular forces |
Delivery of poorly soluble vericonazole (Pfizer, USA) as intravenous solution for systemic fungal infection Delivery of omeprazole (Betafarm, Germany) as tablet for improved bioavailability. Improved delivery of nicotine as sublingual tablet (Pharmacia, Sweden) |
Quantity of drug loading and dosage form design may be limiting factors Challenge of scale up of manufacturing process |
Kanikkannan6 and de Miranda et al.22 |
| Crystal engineering | Controlled crystallization of drugs so as to produce powders with crystal form (crystalline or amorphous), high purity, surface energy- defined particle size distribution and crystal habit by manipulating different solvents or change in the stirring or adding other components |
Delivery of chloramphenicol palmitate as meetastable polymorph for enhanced solubility and absorption Delivery of oxytetracycline using form B polymorph for improved solubility |
Savjani et al.3 | |
| Hot melt extrusion | Process of dissolving poorly soluble APIs into a polymer matrix known to form an amorphous solid dispersion |
Delivery of oleanolic acid for improved dissolution |
Challenge to achieve high drug loading Not suitable for thermosensitive drugs and polymers Limited choices of pharmaceutical-grade polymers and surfactants |
Gao et al.23 |
| Lipid-based microparticulate drug delivery system (solid lipid microparticles) |
Drug delivery using microsized drug carriers with solid lipid matrices such as fatty alcohol, solid wax, fatty acids or glycerides |
Delivery of artemether for improved improved solubility, dissolution permeability and bioavailability delivery of dihydroartemisinin -piperaquine phosphate for malaria |
Potential for interaction of lipid carrier with drug structure or its degradation limited studies on the use of lipids with high melting points The use of temperature may limit the use of thermolabile drugs |
Agubata et al.24 Amarachi et al.25 |
|
The liquisolid technique, as another promising strategy used to circumvent poor solubility, dissolution rate and bioavailability in medicine development, is now gaining wider attention and application as a drug delivery system. From its early introduction about 3 decades ago, much is now being made available by researchers who have explored this area of research in delivering drugs with poor water solubility. An up-to-date understanding of this technique is therefore critical for its use and application in improving the delivery of new and existing challenging drug molecules.
Liquisolid technique: Principle: First discovered and propagated by Spireas in his doctoral thesis and when employed to deliver steroids such as prednisolone, the liquisolid system is an advanced technique of solid oral dosage form for enhancing solubility and dissolution of poorly soluble drugs26,27. It has reportedly been valuable in overcoming the limitations associated with the other approaches for improving drug bioavailability such as hydrophobic agglomeration (common with micronizing), poor stability (associated with salt formation) uncommon technology (peculiar to nanotechnology-driven techniques) and the influence of pH.
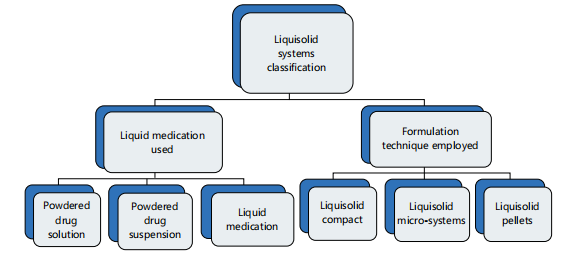
The liquisolid technique has been considered an outgrowth of the powdered solution technology that was used in the delivery of liquid medication28. In its most simplistic definition, it is an approach of drug delivery where in a poorly soluble drug is formulated as liquid medicine but delivered in a solid dosage form. The poorly soluble solid drugs are initially made into liquid medicines by either dispersing or dissolving them in non-volatile solvents and then converted to flowable powders or granules that are non-adherent but compressible. This transition from liquid state to solid powders or granules is made possible through the use of carriers and coating materials. A simple illustration of the classification of the liquisolid system is shown in Fig. 2.
The formulation techniques and the resulting presentation further reveal the variety brought into this delivery system.
Liquisolid compact: These are liquisolid systems that are presented as tablets or capsules. They come as sustained or immediate-release formulations and have the inclusion of other excipients (disintegrants, lubricants, binders, etc.,) that make them readily compacted or encapsulated.
Liquisolid microsystem: These liquisolid systems are presented in multipellets or microsized granules or beads and encapsulated, usually contains adjuvant polyvinylpyrrolidone (PVP).
Theoretical principle: The liquisolid approach to drug delivery is based on the following fundamental pharmaceutics principles:
| • | Principle on flowability and compressibility: A given porous powder mass with acceptable flowability and compressibility can absorb (hold) a limited amount of liquid. This amount of retainable liquid is carrier-specific, coating material-specific and liquid-specific. This implies that the amount of liquid paraffin, for instance, that microcrystalline cellulose (a carrier) can hold and be flowable will be different if the liquid is changed to glycerol or the carrier is replaced with aerosol | |
| • | Based on this principle, in liquisolid systems, drug(s) in the liquid state are converted into apparently dry powder, with acceptable flow and compressible properties by simply introducing the liquid medication (either by blending with or spraying onto) selected excipient carriers and coating materials29 | |
| • | That limit to the amount of liquid the powder mass can hold is a function of its specific surface area and absorptive capacity. Materials having higher values of specific surface area or absorptive capacity can hold a larger volume of the liquid medication. Hence, this principle provides guidance on the selection of powders for use as carriers and coating materials for optimal delivery. It must be mentioned too that, as reported in the literature, the incorporation of binders such as hypromellose or povidone reduced the amount of a blend of carrier-coating agents used to obtain flowable and compressible powder mass29 |
Principle of improving dissolution and solubility: The dissolution rate and solubility of a poorly soluble drug can be improved by increasing its wettability and surface availability to the dissolving medium. Wettability and surface availability can be increased by reducing the contact angle of the drug substance and the solvent using surface-acting agents or employing liquids with better wetting properties. When the drug (to achieve improved wetting) is thus dispersed in a solvent that is water miscible, it enhances faster dissolution and overall solubility of the drug in the aqueous medium. This principle finds its value in providing needed guidance on the choice of appropriate solvent for the optimal formulation of the liquid medicine in liquisolid delivery. Liquid medicine can be presented as a suspension, solution or emulsion of the drug28.
Principle of impact of excipient ratio: The compressibility and mechanical properties of the formulated solid dosage form (compacts or tablets) are dependent on the properties of the excipients (carrier in this case) used and their ratio to the coating material in the formulation. This highlights the importance of the ratio of carrier material to coating material in achieving a liquisolid compact having desirable properties. In literature the ratio of carrier to coating agent of between 10-20 to 1 but recent works have reported even higher ratios even up to 50:130,31.
Basing this technique of development of medicine on such foundational principles of pharmaceutics gives it some merit. Table 2 presents some of these alongside some disadvantages.
Mathematical concepts: To achieve a liquisolid system having the desired flow properties and compressibility, the liquid medication and excipients (the carrier and the coating agents) must be used in the right proportions. Some mathematical concepts are employed for accurate determinations of these proportions as follows.
Spireas and Bolton in 1999 as reported by Tiong and Elkordy32, introduced an empirical method to determine the amount of each excipient to be used in the formulation of the liquisolid compact on the basis that each excipient in a liquisolid system (carrier or coating material) possessed a flowable liquid-retention potential: For the carrier (Φ) and the coating materials (φ). This also holds when using different mass ratio mixes of carrier and coating materials. The values are required to calculate other necessary
| Table 2: | Merits and demerits of the liquisolid system | |||
| Merits of the liquisolid systems | Demerits of liquisolid system |
| Versatility in delivery technique and useful for enhancing solubility of poorly water-soluble drugs, slightly soluble ones and practically insoluble drugs. It can now be applied to even soluble drugs to sustain the release |
In many cases, mainly applicable to drugs that come as low- dose medicines. Drugs with higher dose strength will require a larger amount of liquid for preparation of drug as liquid medication, thus requiring more excipient use with results of very large difficult-to-swallow tablet |
| Relatively cheap and simple technology to use in drug delivery | Seemingly limited choice of dissolving/dispersing solvents Other liquid vehicles are being considered for use |
| It leverages the tableting and encapsulating technology as the popular oral route of administration |
It requires necessary expertise in the application of the mathematical concepts in determining the appropriate quantities of excipients to be used in the formulation |
| It can be easily scalable and hence has good industrial application | The non-volatile solvents used in preparing the liquid medication can negatively influence the mechanical strength of the compacts manufactured |
| Application of technique now expanded to modified delivery system, circumvent photosensitivity reactions in drugs and influence of pH on drug solubility during manufacture The drug formulation though solid is actually a drug in solubilized or well-dispersed liquid form hence allow faster wetting and resultant enhanced dissolution |
parameters of the system. The flowable liquid retention potential of a carrier, for example, is ascertained by gradually incorporating a liquid medication onto its bulk powder of the carrier, blending and then determining the flow properties of the bulk powder. This is repeated until when the optimal flow properties are obtained. The amount of liquid held at that point by the carrier is its flowable liquid retention potential. A particular flow property of significance and being used for such determination is the angle of the slide. The same is determined for the coating material. So the liquid load factor, Lf , the flowable liquid retention potential of the carrier, Φ, the same parameter for the coating material, φ and their mass ratio (R) for each liquid medication is related, according to Spireas by Equation 1 below32:
| (1) |
Secondly, the liquid load factor can affect the compressibility of the powder mass. The maximum liquid a bulk carrier powder can hold while maintaining optimal compressibility is termed the compressible liquid retention potential. This is specific for the carrier and coating agent. Lu et al.33 maintain that the compressible liquid retention potential is readily determined by pactisity. Hence, a valuable parameter of interest becomes compressible liquid retention potential for the carrier as well as the coating material. Equation 2 relates Lf , compressible liquid load factor, to the compressible liquid retention potential for the carrier (Ψ), compressible liquid retention potential for the coating material (ψ) and the mass ratios of the excipients (R) as follows33:
| (2) |
At the combination of carrier and coating material, the amount of liquid medication that can be held by such a system while remaining flowable and compressible becomes dependent on the excipient ratio, R. That excipient ratio (R) is the weight ratio of the carrier (Q) to the coating material (q) in the powder mix as given in this third equation below32:
| (3) |
In line with the third principle of the liuisolid technique, R is related to the compressibility, flow properties and mechanical properties of the liquisolid system hence an optimal value for R is desirable and recommended in literature to be from 10 to 2027,33,34. Recent works show, however, that R can go up to 35, even 50 and still achieve intended formulation outcome30.
The maximum amount of liquid that a given excipient ratio can hold while still maintaining flowability or compressibility is the liquid load factor (Lf). At such an optimal mix, Lf becomes Lo that is the optimal load factor. In mathematical terms, it is the ratio of weight of liquid medication (W) to weight of carrier material (Q) as presented here in Equation 434:
| (4) |
The Lf is also obtainable from the in Equation 1 or 2, whichever one of the two is the lower value. This value of as calculated is not the same as one that is usually determined experimentally because the former does not take into account the amount of drug in the liquid medication which will affect the total amount of liquid that will be available for absorption and adsorption by the carrier/coating material mix35.
So once the weight (W) of liquid medication and the loading factor (Lf) are known, the amount of carrier and coating materials to be used to obtain an optimal mix is readily determined so that the loaded amount of the liquid vehicle would not hinder the flowability and compressibility of the liquisolid system.
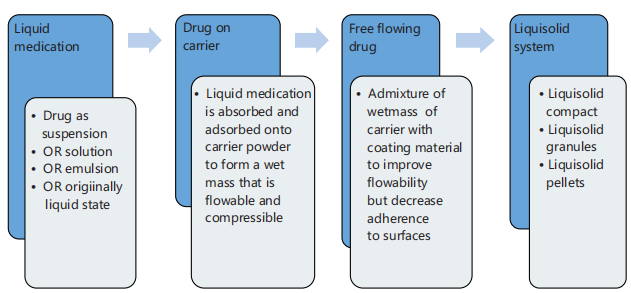
Once the different proportions of the ingredients of the liquisolid system are determined, the procedure for formulating the liquisolid system is followed strictly to obtain the desired results. These steps are well illustrated summarily in Fig. 3.
Formulation considerations of liquisolid dosage forms
Drug selection: The liquisolid technique of drug delivery was set out to solve the pharmaceutical problem of poor solubility. Thus originally, drugs selected for the liquisolid systems usually are those in the Class II and 1V of the Biopharmaceutical Classification System. Drugs in these classes have aqueous solubility challenges that the technique attempts to circumvent. Nevertheless, drugs in the other classes are now being considered when specific applications are intended (e.g., to overcome pH influence or achieve sustained drug release). Even in these later situations, solubility is still a valid concern as it influences the dissolution profile of the final compact and its size.
|
Another formulation consideration in drug selection is the dose and solubility of drugs delivered as a liquisolid system. To achieve a lower final tablet weight, the drug dose (loading dose) is usually of small weight (usually 1-50 mg) because the carriers and the coating materials generally constitute the larger percentage weight of the final tablet presentation. When drug dose is larger, this results in final tablet size and weight being too high for oral compact as the tablet size will be inconvenient for swallowing. Some of the drugs that have been formulated as such includes itraconazole, Olmesartan Medoxomil and Meloxicam36-38.
Liquid vehicle (non-volatile solvent): This component is used to dissolve/disperse the solid drug to form liquid medicine. The considerations for its use, beyond safety for human consumption and inertness, are solubility capacity, low viscosity, non-volatility and water miscibility. The intended application of the liquisolid system is also considered. It must be noted that the degree to which the liquid vehicle possesses these properties gives indication to how it will influence the final compact. For instance, liquid vehicle with low solubility capacity will have less molecular dispersion of the drug in it. This will translate to the use of more liquid vehicle to form the compact and larger carrier and coating agent used to take up the liquid and resulting tablet size likely to be large. Formulations with vehicles having low solubility capacity may favor a sustained release application. Commonly used liquid vehicles are glycerin, polyethylene glycol, PEG 200, 400 and 600, polysorbate (Tween) 20 and 80. They can be used singly or in combinations as co-solvents. The liquid vehicle even adds to the compactness of a liquisolid system likely due to the presence of hydrogen bonds and the degree of drug solubility or dispersibility in the liquid is directly related to percentage dissolved during dissolution studies33,39 . Although, originally, water miscible liuid vehicles were the desired in formulation using liquisolid systems as designed by Spireas non-water-miscible vehicles (such as castor oil and cremophor EL have now been applied in modern use40.
Carriers: The carriers in liquisolid systems are used to absorb the liquid medicine converting it to solid relatively flowable powders. To select a given material as a carrier, the following properties are considered:
| • | Specific surface area (SSA): This is the surface area possessed by a unit mass of the carrier. Higher value of SSA contributes to higher absorptive capacity to the carrier, reducing the quantity that will be used in each formulation and by extension the final compact size and weight | |
| • | Absorptive capacity: This is the amount of liquid medicine that can be held within a unit mass of the carrier. The SSA, nature of a given material and its surface characteristics determine its absorptive capacity |
Thus the ratio of liquid phase to the carrier material had been reported to have an effect on the mechanical properties (hardness, friability, disintegration of tablet and tablet height) of the resulting liquisolid compacts formed29. Higher SSA and absorptive capacity are desirable properties in carriers beyond safety and inertness, to checkmate the final tablet weight. Commonly used examples of carriers for liquisolid systems and some of their are found in Table 3.
Applications of the liquisolid systems: The use of liquisolid technique has been reported in many works in the area of enhancing drug bioavailability through improved dissolution and solubility. This is not surprising as this is what the technique, as developed by Spireas was set out to achieve. Recent works, however, have expanded the techniques to preparing sustained release drug delivery, to overcome the influence of variation in pH for drugs whose dissolution rates are pH dependent and for photoprotection of light-sensitive drugs.
Liquisolid techniques as a delivery strategy to enhance dissolution, solubility and bioavailability of medicines: The bioavailability of a drug is related to its dissolution rate, aqueous solubility and intestinal permeability. Spireas and Sadu27 in his invention of the liquisolid technique was originally to improve drug dissolution and solubility, hence by extension bioavailability. Several authors have since applied the technique to achieve enhanced dissolution, solubility and bioavailability27,32,34,36-38. For example, Vranikova et al.29 applied liquisolid technique to deliver rosuvastatin (a hypolipidemic drug) using both the spray method and simple mixing method to incorporate the formulated liquid medication into the carrier-coating powder mass. The results showed that in both methods of incorporation, about 80% of the rosuvastatin was released within 5 min from the formulated liquisolid tablets. These researchers also found that rosuvastatin liquisolid tablets formed had hardness and friability that were affected by the ratio of the liquid medication to the carrier used in the formulation. They highlighted that the mode of introduction of the liquid medication was a factor to consider during the preparation of liquisolid systems as compacts made from the spray drying process of incorporation produced compacts with higher mechanical properties, better micropolitics and dosage uniformity.
| Table 3: | Commonly used carriers for liquisolid delivery and their peculiar characteristics41 | |||
| Classes of carriers or coating material |
Specific examples | Specific surface area (m 2/g) |
Absorption capacity (mL/g) |
| Polysaccharides | Starches (tuber starches) | 0.6-4.35 | 0.96-1.5 |
| (corn starches) | 1.22-1.26 | ||
| Microcrystalline cellulose(Avicel PH 101) | 1 | 0.04-0.08 | |
| Amorphous cellulose | 12-22 | ||
| Lactose | 0.35 | ||
| Sugar alcohol | Sorbitol | 0.37-1.05 | |
| Phosphates | Fujicalin® (anhydrous dibasic calcium phosphate) |
27~40 | 1.2 |
| Dicalcium phosphate dihydrate (DCPD) Florite® |
0.3 | 1-1.3 | |
| Silicates | Neusilin (magnesium aluminometasilicates) Aerosil®200 |
300 | 3.4 |
| Microsilica | 200 | 1.7 | |
| 78 | 2.9 | ||
| Ordered mesoporous silicates | 1030-1500 | 7.8 |
Similarly, Thakkar et al.36 attempted to enhance the bioavailability of itraconazole (a potent antifungal) using the liquisolid technique (PEG 600 as the non-volatile liquid, Aerosil 200 as coating material and branded microcrystalline cellulose [Alfacel PH 200] as carrier). Their optimized formulation (which had 10 mg drug in 150 mg liquid) had over 60% dissolution in less than 50 minutes, unlike the marketed brand which was about 40% in the same duration. They also noted that using a higher quantity of dissolution liquid (beyond 150 mg) in the formulations had no significant improvement in the percentage dissolution because saturation in solubility had occurred.
Table 4 below highlights several other examples of published research that has applied liquisolid technique with remarkable success for improved solubility, enhanced dissolution and drug bioavailability enhancement.
About three different mechanisms have been put forward as possible ways that the liquisolid systems bring about enhanced dissolution and bioavailability:
| • | By increasing the available effective surface area of the drug for dissolution: The drug is molecularly dispersed and held in that state onto the surface of the carrier and coating materials during blending. This surface area is then made available, on the administration of the dosage form, to the gastrointestinal fluid for faster dissolution and subsequent absorption42,43. Saeedi et al.42 even established in their work on dissolution enhancement of indomethacin by liquisolid system, that the fraction of the drug molecularly dispersed in the liquid medication of liquisolid systems was directly proportional to dissolution rate of the indomethacin compact |
| Table 4: | Published works on liquisolid delivery systems showing enhanced dissolution, solubility and bioavailability | |||
| Drug | Non-volatile liquid used |
Carrier/coating material |
Improvement over marketed brand (%) |
Presentation/ application |
Reference | |||
| Valsartan | Propylene glycol |
Avicel/aerosil 200 |
Over 15% in dissolution efficiency |
As liquisolid compact |
Chella et al.44 | |||
| Rosuvastatin | PEG 400 | Neusilin US2/Aerosil 200 |
Fast disintegration and enhanced dissolution profiles |
As liquisolid tablet | Vraníková et al.29 | |||
| Ritonavir | PEG 400 | MCC/crospovidone | Improved dissolution by over 40% |
As liquisolid pellets | de Espíndola et al.45 | |||
| Telmisartan | Transcutol HP | Avicel PH02/Aerosil 200 |
Significant improvement | Liquisolid compacts | Chella et al.46 | |||
| Efavirenz | Transcutol HP | Neusilin US2 and corn starch/Aerosil |
Improved dissolution profile with almost 100% release in 60 min |
Liquisolid tablet | Jaydip et al.47 | |||
| Olmesartan Medoxomil |
Acrysol El 135 (Polyoxyl 35 castor oil) |
Avicel PH 102, Fujicalin and Neusilin/Aerosil |
Significant higher drug release rates |
Liquisolid compact | Prajapati et al.37 | |||
| Olanzapine | Kolliphor EL | Avicel/ Aerosil | Formulations showed good/excellent flow properties and compressibility AUC of optimized liquisolid formulation was higher than marketed tablet |
Liquisolid tablet |
Korni and Gonugunta48 | |||
| Meloxicam | PEG 400 | Avicel PH102/Aerosil | Higher dissolution with more than 80% drug release within 10 min |
Liquisolid tablets | Dias et al.38 | |||
| Fexofenadine hydrochloride |
Propylene glycol or Cremophor ®E L |
Aerosil® 200 Avicel® PH102/ |
Increased oral drug bioavailability by 62% and reduced Tmax to 2.16 hrs |
Liquisolid tablets | Yehia et al.49 | |||
| Itraconazole | PEG 600 | Afacel PH200/ Aerosil |
Higher drug dissolution, Cmax an AUC |
Liquisolid compact | Thakkar et al.36 | |||
| Prednisolone | PEG 400, glycerin, propylene glycol |
MCC/Silica | Higher dissolution rate with enhanced bioavailability |
Liquisolid compact | Spireas and Sadu27 |
|||
| Indomethacin | PEG 200, glycerin | Avicel PH101/ Nano-sized amorphous silica |
Liquisolid formulations exhibited significantly higher drug dissolution rates than directly compressed tablet |
Liquisolid compact | Saeedi et al.42 | |||
| Famotidine | Propylene glycol | Avicel® PH 102/ Aerosil® 200 |
Optimized liquisolid formula had 39% higher release than directly compressed tablets during the first 10 min |
Liquisolid tablet | Fahmy and Kassem50 |
|||
| Carbamazepine | PEG 200, PEG 400 | MCC or Lactose/ Nano-sized amorphous silica and PVP or HPMC or PEG35000 added to reduce quantity of carrier-coating mixture used |
Liquisolid formulations containing PVP showed significantly higher drug dissolution rates over that prepared as directly compressed compacts technique Improved dissolution as carrier- coating ratio reduced from 20 to 10 but decreased when reduced to 5 10 |
Liquisolid compact | Javadzadeh et al.34 | |||
| Furosemide | Castor oil, cremophor EL and PEG 400 in ratio 1:6:3 |
(Avicel PH101) and coating materials (Aerosil 200) |
2-folds increase in drug release |
SNEDDS delivered as liuisolid tablets |
Dalal et al.40 | |||
| Atorvastatin | PEG 400 | Avicel PH 101 OR Avicel PH 102 OR Neusilin US2 as carriers and Aerosil 200 as coating material |
Optimized liquisolid tablets showed higher dissolution compared to marketed tablets |
Liquisolid tablets | Windriyati et al.51 | |||
| Clopidogrel | Propylene glycol and water in ratio 2:1 |
Maize starch, microcrystalline cellulose/colloidal silicondioxide |
Formulation showed higher solubility in HCl buffer pH 2 |
Liquisolid tablet | Ali et al.52 | |||
| • | Drug is presented in a dissolved state: Drugs administered orally undergo some steps (disintegration, deaggregation, then dissolution) before absorption in the gastrointestinal tract. But when the drug is already in the dissolved state in the liquid vehicle, adsorbed onto the carrier and coating material, these preliminary steps are readily passed and the dissolution rate is enhanced for improved bioavailability | |
| • | Improved wetting properties of the liquisolid compacts: A key factor that contributes to poor dissolution of solid oral compacts is their poor wettability in the dissolution medium has been explained to be a function of the high interfacial tension between the two phases. In liquisolid technique of drug delivery, the non-volatile liquid used to prepare the delivery system decreases the interfacial tension between the compact surface and the dissolution medium thus resulting in better wetting properties of the final solid dosage form as a first step towards enhanced dissolution42. Vranikova et al.29 reported this as one mechanism used for enhanced dissolution in their work involving rosusvastatin |
As a means to minimize influence of pH variation on dissolution: Many drugs are weak acids, bases, or their salts. Hence their solubility and subsequent absorption in the gastrointestinal tract (GIT) is influenced by their ionization constant (pKa) and the pH of the local dissolving environment. After enteral administration, drugs move in the fluid secreted along the gastrointestinal tract. The pH of the gastrointestinal environment varies from the mouth to the large intestine and a large extent, this is a function of whether it is in a fasted or fed state. Thus these changing pH values in the GIT will affect the dissolution, solubility and bioavailability of these ionizable drugs as they gradually coast along in the gastrointestinal fluids. This variation in a person throughout his regimen and between persons could affect, the onset of action, therapeutic outcomes and expressed untoward effects. Drugs formulated via the liquisolid delivery system have now been reported to effectively minimize the impact of varying pH on drugs whose dissolution and solubility are affected by changing pH at the site of absorption.
El-Hammadi and Awad35 investigated the use of liquisolid technique to reduce the effect pH variations will have on the release of loratadine, employing propylene glycol, Avicel PH 102 and Aerosil 200 as non-volatile liquid, carrier and coating materials respectively. The formulated liquisolid systems were evaluated in 3 dissolution media, each of different pH (pH 1.2, 2.5 and 5). Results showed that, generally, the process of drug release becomes slower as the pH value of dissolution medium increased from 1 to 5. The optimized formulation of liquisolid systems showed significantly enhanced dissolution and drug release than the marketed brand at a higher pH of the dissolution medium. One mechanism used to explain this application of liquisolid technique is that liquid used to dissolve or disperse the drug in the formulation influences the saturation solubility thereby enabling maintenance of a good concentration gradient when in a dissolution medium of different pH thus maintaining dissolution35.
Table 5 presents a list of some of the successful claims in literature on this application of liquisolid technique in minimizing pH effect.
Liquisolid technique as a means to achieve photostability of photosensitive drugs: Photodegradation, loss of potency and subsequent likely production of toxic degradation molecules could readily result when photosensitive drugs are exposed to ultraviolet light. These consequent potential adverse effects can be devastating and hazardous to health. The liquisolid technique is gradually finding useful applications in photoprotection, as mentioned in the literature, to provide solution to the longstanding challenge of drug sensitivity to light. Khames54 for example, had reported the evaluation of amlodipine (a photosensitive antihypertensive and antianginal drug) formulated as liquisolid tablet (using propylene glycol as liquid vehicle and Avicel /amorphous silicon or nano-sized TiO2 as carrier/coating material respectively) under the influence of ultraviolet light to see the degree of photoprotection conferred on it by the technique. The optimized liquisolid system inhibited photodegradation under different light energies.
| Table 5: | List of examples of drugs that have been delivered as liquisolid systems to minimize the effect of pH variation | |||
| Drug | Non-volatile liquid used |
Carrier/coating material |
Effect of pH variation |
Presentation/ application |
Reference |
| Loratadine (antihistamine, used in rhinitis, urticaria etc,) |
Propylene glycol | MCC/Silica | Dissolution of liquisolid tablets were significantly higher than popular marketed brand at higher pH of dissolution medium Liquisolid formulation less affected by pH variation in comparison with the directly compressed tablets and Clarityn® |
liquisolid tablets | El-Hammadi and Awad35 |
| Mosapride citrate (gastroprokinetic: Useful in dyspepsia and acid reflux) |
Glycerol | Avicel PH 102, lactose DC as lactose DC as carriers/Aerosil 200 as coating material |
Minimize the effect of pH variation on drug release along the gastrointestinal tract using bio-relevant media |
Liquisolid tablets | Badawy et al.53 |
Khazim et al.31 recently carried out similar research on Nimlodipine employing propylene glycol and Avicel, as nonvolatile solvents and carriers respectively but colloidal silicon dioxide, Cab-O-sil and titanium dioxide as the different coatings materials. They reported significantly improved photoprotection by the liquisolid systems formulated.
But how is photoprotection possible with the liquisolid technique? Researchers propose that the photoprotection by this strategy is based on photoprotective capacity of coating material (silicon dioxide) used in liquisolid system, having a high refractive index and diffraction for the light waves of different energies and wavelength so that the drug is protected from the effect of the light. Khames54 also noted that the photoprotective effect was inversely proportional to the excipients ratio (R). This application opens an opportunity to employ the liquisolid technique, with the coating material having good refractive and diffractive capacities as a good alternative to the conventional coating process. It will thus reduce the additional production step and cost of coating photosensitive solid dosage forms. Table 6 shows details of some photosensitive drugs that have been protected using the liquisolid technique.
liquisolid system used to achieve sustained release formulations: Another significant area of application of the liquisolid technique has been in modified-release formulations, especially sustained release. Hydrophobic carriers (e.g., ethyl cellulose, Eudragit® RL) when used in liquisolid compacts can confer reduced wetting on liquisolid formulation, thus modifying the drug’s release rate from the compact. If such carriers are combined with drug release retardants (such as hydrocolloids like hydroxypropyl methyl cellulose, HPMC) in matrix systems, the influence and degree of release modification are heightened to achieve prolonged or sustained drug release. Several authors have employed this and have reported success. Pavani et al.30 for example attempted to produce sustained release formulation of trimetazidine dihydrochloride liquisolid tablets. They employed ethyl cellulose, Eudragit® L 100 and
| Table 6: | Some published works on application of the liquisolid system to achieve protection for photosensitive drugs | |||
| Drug | Non-volatile liquid used |
Carrier/coating material |
Degree of photoprotection |
Presentation/ application |
Reference |
| Nimodipine | propylene glycol | Avicel/colloidal silicon dioxide, Cab-O-sil and titanium dioxide |
Liquisolid significantly improved photo protection |
Liquisolid tablet | Khazim et al.31 |
| Amlodipine | propylene glycol | Avicel/amorphous silicon or nano-sized TiO2 |
Inhibited the photodegradative effect of different light energies in all prepared liquisolid formulations |
Liquisolid tablet | Khames54 |
Eudragit® RS 100 as retarding agents along with Avicel PH 200 as the carrier, coating material (Aerosil 200) and polysorbate 80 as the nonvolatile liquid to prepare liquisolid tablets. In the dissolution studies of the formulated liquisolid tablets as well as the marketed brands carried out in media of pH 1.2 for first 2 hrs and then in pH 7.4 for the next 12 hrs, they found out that while all the formulations showed potential, the one with Eudragit L100, optimized formulation, sustained the drug release significantly better than the marketed brand and followed a zero order drug release kinetics. That release profile was maintained even after storage for 6 months at temperature of 40±2°C and relative humidity of 75±25%. The possible mechanism of prolonged drug release here is by poor wetting which delays the disintegration stage for drug release to occur30,52.
Ali et al.52 in their work on clopidrogel formulated sustained release oral liquisolid tablets using hydrophilic carriers HPMC and PVP. The HPMC was found to sustain the release of clopidrogel for over 4 hrs in the 0.1 HCl dissolution medium and the sustaining release in the liquisolid compact was dependent on the quantity of the hydrophilic polymers employed. The mechanism as proposed by the researchers is by the formation of a gel by the hydrophilic polymer on absorption of gastrointestinal fluid thus retarding the amount and rate of release of the drug from the core rather than simply inhibiting wetting of the matrix. This proposed mechanism alligns with what other researchers working with hydrocolloids have used to explain the drug release mechanism from such polymers whether natural or modified biopolymers55,56.
Other factors that have been suggested to contribute to achieving sustained drug release in liquisolid systems include the use of carrier with lower specific surface area; ratio of drug in liquid vehicle; the ratio of carrier, retardant and coating material in the formulations investigated; choice of liquisolid vehicle for modulating the drug release rate and drug concentration in vehicle, where high concentration sustains release because at a higher drug concentration, the drug tends to precipitate within the dissolution medium42,57. Table 7 below presents some published literature on drugs that have been delivered as sustained release using the liquisolid system.
Limitations of the liquisolid system: Despite the current wide application of the liquisolid drug delivery technique in improving dissolution of poorly soluble drugs, limiting pH influence on drug release, protecting light-sensitive drugs and achieving sustained release, there are reported peculiar limitations. These are discussed in this section; using the conventional infrastructure for tableting gives the liquisolid techniques potential for industrial application. However, the scale-up of the procedure has been hampered by the challenge of poor and unpredictable flowability and compressibility of the produced liquisolid powder mixtures. This is reflected in the mechanical properties of the compacts usually produced. Another problem is that of mixing, especially when a viscous solvent is used to prepare the liquid medication, or when a small quantity of such liquid is to be incorporated into a large mass of carrier material.
Achieving complete mixing in such a situation may be a real challenge. At Industrial quantities, the challenge of free flow of the liquisolid mix may result in nonuniformity of doses. Another challenge could be in the method of incorporation of the liquid medication onto the carrier powder mass which significantly contributes to compaction behavior of liquisolid systems. In the work of Aleksic et al.60, who investigated the effect of formulation variables (liquid content, spray air pressure and liquid feed rate) on the compaction properties of liquisolid systems, they found that liquisolid system prepared using fluid bed processor gave better compaction behaviour while taking up higher liquid medication.
| Table 7: | List of published works of sample drugs showing application of liquisolid technique in achieving sustained release | |||
| Drug | Nonvolatile liquid | Carrier/coating agent | Application | Presentation | References |
| Trimetazidine dihydrochloride |
Polysorbate 80 | Ethyl cellulose, Eudragit and RS100/Aerosil 200 |
Sustain release | Liquisolid tablets | Pavani et al.30 |
| Theophylline | Non-volatile co-solvent |
HPMC | Presence of non-volatile co-solvent was critical for prolonging drug release. The compared with the directly compressed tablets |
Liquisolid tablets | Nokhodchi et al.58 |
| Propranolol HCl | Polysorbate 80 | Eudragit RL 100 or RS as the carrier/silica as coating material |
Sustained release polymer chains coalesced better, resulting in a fine polymer network with lower porosity and higher tortuosity. liquisolid formulation followed zero order |
As sustained-release liquisolid tablets |
Javadzadeh et al.34 |
| Arthemeter- Lumefanntrine |
Precirol® ATO 5/ Transcutol® HP Also, tallow fat/ Transcutol® HP optimizedsystems |
MCC/colloidal silicone dioxide |
Tablet compacts formulated with Precirol® ATO 5/ Transcutol® HP-AL4 achieved higher LUM release in simulated intestinal fluid (84.32%) than tallow fat/ ranscutol® HP-BL3 |
As sustained-release liquisolid compact |
Nnamani et al.59 |
| Tramadol | Propylene glycol | Avicel PH 102/Aerosil 200 but HPMC(K4M) used as release retardant |
Sustained release is dependent on percentage of HPMC in the formulation and release kinetics Korsmeyer -Peppas and is non Fickian release. Sustained longer than that of marketed tablets |
Liquisolid compact |
Karmarkar et al.57 |
| Clopidogrel (antiplatelet) |
Propylene glycol and water (2:1) |
Polyvinyl pyrollidone, HPMC/ |
Formulation achieved sustained release over that prepared by the direct compression |
Liquisolid compact | Ali et al.52 |
Achieving a balance between good flowability and compressibility of liquid drug powder mixture can be challenging. True, the established mathematical equations for calculation of the flowable and compressible mixture can be extrapolated for use. This can be technically demanding and cumbersome requiring good skill in formulation to achieve success. Moreover, the value for liquid retention potential and the liquid compressible potential are determined by repeated experimentation and measurements which may be cumbersome. Besides, the values as determined are different from the experimental value that will eventually be in use because the earlier determination did not incorporate the drug yet. More so there is a challenge of possible squeezing-out effect of the liquid medication during compression of the liquisolid powder mix. One way this is circumvented is the formulation of capsules in place of compressed tablets.
Application to drugs with small doses and limited amounts of liquid medication: Liquisolid technique was originally for application to low-dose medicines. The drug dose determines the amount of nonvolatile solvent used to dissolve or disperse it and will eventually translate to the size of the compact prepared.
Thus applying it to formulate high-dose drugs is a limitation as it portends larger size compact which may be impractical. Rokade et al.28 reported that once the therapeutic dose of a drug is greater than 50 mg, a low-level hydrophilic carrier and coating agent are used in the liquisolid technique. Higher drug doses will require a larger amount of carrier to achieve liquisolid powder mass that is free-flowing. This can result in a tablet weight that is more than 1 g and difficult to swallow61.
To overcome this limitation of suitability for delivering low therapeutic doses, the improvement pellets-have been investigated and used with remarkable success. The liquisolid pellet combines the concepts from the liquisolid technology and pelletization technology. It has the advantage of accommodating higher therapeutic doses of drugs for delivery than the liquisolid compact besides other benefits such as being a multi-particulate system over single-unit dosage forms of liquisolid compacts, better distribution along the gastrointestinal tract with enhanced bioavailability45,62. One possibility for achieving the use of a higher dose is the choice and amount of coating agent used. de Espindola et al.45 used crosspovidone at 30% concentration and maintained an R value (that is, the ratio of carrier to coating material in the formulation) of less than 1 and achieved liquisolid pellets of high drug dose and enhanced dissolution.
The release kinetics in liquisolid systems still have room for improvement and will need further studies. Some published studies reveal that classic liquisolid technique did not significantly affect the drug dissolution profile over conventional tablets as was seen in the advanced form, the liquiground. This interesting improvement on the liquisolid system in terms of improved dissolution rate is achievable by the liquiground technique-a combination of the liquisolid technique and the co-grinding technologies. Azharshekoufeh et al.63 applied this liquiground technique to deliver glibenclamide with significantly improved dissolution rate and enhanced bioavailability. This was possible because particle size reduction achieved by co-grinding of liquid medication was more effective in enhancing the dissolution rate of glibenclamide than the simple implementation of the liquisolid technique.
CONCLUSION AND FUTURE POSSIBILITIES
The liquisolid system has proven to be a versatile technology in drug delivery with improvements in drug release, overcoming poor solubility and dissolution while not altering chemical properties of the drug in the formulations. This review has presented the underlying pharmaceutics principles of the technique, provided ample examples of published works on the application in different areas of drug delivery and identified limitations associated with the formulation strategy. The current advancements to circumvent its presently identified limitations are welcomed to keep improving this highly invaluable technique expanding in applications to drugs of higher doses and to take up higher amounts of liquid medication. Also noteworthy is the fact that the combination of this liquisolid technology with other existing technologies brought about improvements in it. As more application is expected in the future with its combination with existing pharmaceutical technologies, formulation scientists can maximize the potential provided by this technique for optimal therapeutic outcomes.
SIGNIFICANCE STATEMENT
This work provides the underlying principles and scientific insight into published works that used the liquisolid technology for drug delivery and explores its current applications. This piece revealed that although the liquisolid technique was originally designed to enhance dissolution and bioavailability of poorly soluble drugs, at present, it has been applied with published examples in three other areas: Modified drug delivery (e.g., sustained releases), minimizing pH influence in drug release and in the protection of light-sensitive medicines. An understanding of the foundational principles of this technique approaches to achieve them, its limitations and its future possibilities are vital for the use of this relatively low-cost yet versatile pharmaceutical technique to design and develop modern innovative drug strategies for better therapeutic outcomes, patient acceptance and medicine availability.
REFERENCES
- Nagaich, U., 2018. Pharmaceutical applications of liquisolid technique. J. Adv. Pharm. Technol. Res., 9.
- Radke, R. and N.K. Jain, 2022. Enhancement of solubility and bioavailability of BCS class-II Ambrisentan: In vitro, in vivo and ex vivo analysis. Int. J. Appl. Pharm., 14: 67-74.
- Savjani, K.T., A.K. Gajjar and J.K. Savjani, 2012. Drug solubility: Importance and enhancement techniques. Int. Scholarly Res. Not., 2012.
- Vemula, V.R., V. Lagishetty and S. Lingala, 2010. Solubility enhancement techniques. Int. J. Pharm. Sci. Rev. Res., 5: 41-51.
- Varandal, A.B., D.D. Magar and R.B. Saudagar, 2013. Different approaches toward the enhancement of drug solubility: A review. J. Adv. Pharm. Educ. Res., 3: 415-426.
- Kanikkannan, N., 2018. Technologies to improve the solubility, dissolution and bioavailability of poorly soluble drugs. J. Anal. Pharm. Res., 7.
- Bhalani, D.V., B. Nutan, A. Kumar and A.K.S. Chandel, 2022. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines, 10.
- Samineni, R., J. Chimakurthy and S. Konidala, 2022. Emerging role of biopharmaceutical classification and biopharmaceutical drug disposition system in dosage form development: A systematic review. Turk. J. Pharm. Sci., 19: 706-713.
- Zhang, X., H. Xing, Y. Zhao and Z. Ma, 2018. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics, 10.
- Patil, S.K., S.W. Kalpesh, B.P. Venkatesh, M.A. Anup and T.B. Dheeraj, 2011. Strategies for solubility enhancement of poorly soluble drugs. Int. J. Pharm. Sci. Rev. Res., 8: 74-80.
- Kalepu, S. and V. Nekkanti, 2015. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B, 5: 442-453.
- Jornada, D.H., G.F. dos Santos Fernandes, D.E. Chiba, T.R.F. de Melo, J.L. dos Santos and M.C. Chung, 2016. The prodrug approach: A successful tool for improving drug solubility. Molecules, 21.
- Wu, R., X. Mei, Y. Ye, T. Xue and J. Wang et al., 2019. Zn(II)-curcumin solid dispersion impairs hepatocellular carcinoma growth and enhances chemotherapy by modulating gut microbiota-mediated zinc homeostasis. Pharmacol. Res., 150.
- de Sá, I.S., A.P. Peron, F.V. Leimann, G.N. Bressan and B.N. Krum et al., 2019. In vitro and in vivo evaluation of enzymatic and antioxidant activity, cytotoxicity and genotoxicity of curcumin-loaded solid dispersions. Food Chem. Toxicol., 125: 29-37.
- Choi, J.S., N.H. Cho, D.H. Kim and J.S. Park, 2019. Comparison of paclitaxel solid dispersion and polymeric micelles for improved oral bioavailability and in vitro anti-cancer effects. Mater. Sci. Eng.: C, 100: 247-259.
- Arango-Ruiz, Á., Á. Martin, M.J. Cosero, C. Jiménez and J. Londoño, 2018. Encapsulation of curcumin using supercritical antisolvent (SAS) technology to improve its stability and solubility in water. Food Chem., 258: 156-163.
- Shen, H. and M. Zhong, 2006. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J. Pharm. Pharmacol., 58: 1183-1191.
- Agubata, C.O., I.T. Nzekwe, N.C. Obitte, C.E. Ugwu, A.A. Attama and G.C. Onunkwo, 2014. Effect of oil, surfactant and co-surfactant concentrations on the phase behavior, physicochemical properties and drug release from self-emulsifying drug delivery systems. J. Drug Discovery Dev. Delivery, 1.
- Xie, S., L. Zhu, Z. Dong, X. Wang, Y. Wang, X. Li and W. Zhou, 2011. Preparation, characterization and pharmacokinetics of enrofloxacin-loaded solid lipid nanoparticles: Influences of fatty acids. Colloids Surf. B: Biointerfaces, 83: 382-387.
- Chaturvedi, P. and P. Sharma, 2024. A review on microencapsulation as method of drug delivery. BIO Web Conf., 86.
- Han, X., C. Ghoroi, D. To, Y. Chen and R. Davé, 2011. Simultaneous micronization and surface modification for improvement of flow and dissolution of drug particles. Int. J. Pharm., 415: 185-195.
- de Miranda, J.C., T.E.A. Martins, F. Veiga and H.G. Ferraz, 2011. Cyclodextrins and ternary complexes: Technology to improve solubility of poorly soluble drugs. Braz. J. Pharm. Sci., 47: 665-681.
- Gao, N., M. Guo, Q. Fu and Z. He, 2017. Application of hot melt extrusion to enhance the dissolution and oral bioavailability of oleanolic acid. Asian J. Pharm. Sci., 12: 66-72.
- Agubata, C.O., I.T. Nzekwe, A.A. Attama, C.C. Mueller-Goymann and G.C. Onunkwo, 2015. Formulation, characterization and anti-malarial activity of homolipid-based artemether microparticles. Int. J. Pharm., 478: 202-222.
- Amarachi, C.S., A.A. Attama and G.C. Onunkwo, 2022. Assessment of the anti-malarial properties of dihydroartemisinin-piperaquine phosphate solid lipid-based tablets. Recent Adv. Anti-Infect. Drug Discovery, 17: 103-117.
- Tayel, S.A., I.I. Soliman and D. Louis, 2008. Improvement of dissolution properties of carbamazepine through application of the liquisolid tablet technique. Eur. J. Pharm. Biopharm., 69: 342-347.
- Spireas, S. and S. Sadu, 1998. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int. J. Pharm., 166: 177-188.
- Rokade, M., P. Khandagale and D. Phadtare, 2018. Liquisolid compact techniques: A review. Int. J. Curr. Pharm. Res., 10: 1-5.
- Vraníková, B., J. Gajdziok and D. Vetchý, 2015. Modern evaluation of liquisolid systems with varying amounts of liquid phase prepared using two different methods. BioMed Res. Int., 2015.
- Pavani, E., S. Noman and I.A. Syed, 2013. Liquisolid technique based sustained release tablet of trimetazidine dihydrochloride. Drug Invent. Today, 5: 302-310.
- Khazim, M.E., H.J. Hasan, N.M. Hanoon and H.A.H. Al-Sa’idy, 2021. Improvement of the photostability of nimodipine by using liquisolid compacts technique. Univ. Thi-Qar J. Sci., 8: 43-51.
- Tiong, N. and A.A. Elkordy, 2009. Effects of liquisolid formulations on dissolution of naproxen. Eur. J. Pharm. Biopharm., 73: 373-384.
- Lu, M., H. Xing, J. Jiang, X. Chen, T. Yang, D. Wang and P. Ding, 2017. Liquisolid technique and its applications in pharmaceutics. Asian J. Pharm. Sci., 12: 115-123.
- Javadzadeh, Y., B. Jafari-Navimipour and A. Nokhodchi, 2007. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine). Int. J. Pharm., 341: 26-34.
- El-Hammadi, M. and N. Awad, 2012. Investigating the use of liquisolid compacts technique to minimize the influence of pH variations on loratadine release. AAPS PharmSciTech, 13: 53-58.
- Thakkar, H.P., D. Vasava, A.A. Patel and R.D. Dhande, 2020. Formulation and evaluation of liquisolid compacts of itraconazole to enhance its oral bioavailability. Ther. Delivery, 11: 83-96.
- Prajapati, S.T., H.H. Bulchandani, D.M. Patel, S.K. Dumaniya and C.N. Patel, 2013. Formulation and evaluation of liquisolid compacts for olmesartan medoxomil. J. Drug Delivery, 2013.
- Dias, R.J., S. Ranjan, K.K. Mali, V.S. Ghorpade and V.D. Havaldar, 2017. Liquisolid compacts of meloxicam: In-vitro and in-vivo evaluation. Egypt. Pharm. J., 16: 112-120.
- Leaner, C. and J. Dressman, 2000. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm., 50: 47-60.
- Dalal, L., A.W. Allaf and H. El-Zein, 2021. Formulation and in vitro evaluation of self-nanoemulsifying liquisolid tablets of furosemide. Sci. Rep., 11.
- Schlack, H., A. Bauer-Brandl, R. Schubert and D. Becker, 2001. Properties of Fujicalin®, a new modified anhydrous dibasic calcium phosphate for direct compression: Comparison with dicalcium phosphate dihydrate. Drug Dev. Ind. Pharm., 27: 789-801.
- Saeedi, M., J. Akbari, K. Morteza-Semnani, R. Enayati-Fard, S. Sar-Reshteh-dar and A. Soleymani, 2011. Enhancement of dissolution rate of indomethacin: Using liquisolid compacts. Iran. J. Pharm. Res., 10: 25-34.
- Pas, T., S. Verbert, B. Appeltans and G. van den Mooter, 2020. The influence of crushing amorphous solid dispersion dosage forms on the in-vitro dissolution kinetics. Int. J. Pharm., 573.
- Chella, N., N. Shastri and R.R. Tadikonda, 2012. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm. Sin. B, 2: 502-508.
- de Espíndola, B., A.O. Beringhs, D. Sonaglio, H.K. Stulzer, M.A.S. Silva, H.G. Ferraz and B.R. Pezzini, 2019. Liquisolid pellets: A pharmaceutical technology strategy to improve the dissolution rate of ritonavir. Saudi Pharm. J., 27: 702-712.
- Chella, N., N. Narra and T.R. Rao, 2014. Preparation and characterization of liquisolid compacts for improved dissolution of telmisartan. J. Drug Delivery, 2014.
- Jaydip, B., M. Dhaval, M.M. Soniwala and J. Chavda, 2020. Formulation and optimization of liquisolid compact for enhancing dissolution properties of efavirenz by using DoE approach. Saudi Pharm. J., 28: 737-745.
- Korni, R.D. and C.S.R. Gonugunta, 2024. Olanzapine liquisolid tablets using kolliphor el with improved flowability and bioavailability: In vitro and In vivo characterization. Turk. J. Pharm. Sci., 21: 52-61.
- Yehia, S.A., M.S. El-Ridi, M.I. Tadros and N.G. El-Sherif, 2015. Enhancement of the oral bioavailability of fexofenadine hydrochloride via cremophor® el-based liquisolid tablets. Adv. Pharm. Bull., 5: 569-581.
- Fahmy, R.H. and M.A. Kassem, 2008. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm., 69: 993-1003.
- Windriyati, Y.N., M. Badriyah, D.A. Kusumaningtyas and R.L. Riesmalia, 2020. Liquisolid tablets formulation of atorvastatin calcium using polyethylene glycol 400 as solvent and some carrier materials. Indones. J. Pharm., 31: 305-311.
- Ali, B., A. Khan, H.S. Alyami, Majeed Ullah, A. Wahab, M. Badshah and A. Naz, 2021. Evaluation of the effect of carrier material on modification of release characteristics of poor water soluble drug from liquisolid compacts. PLoS ONE, 16.
- Badawy, M.A., A.O. Kamel and O.A. Sammour, 2016. Use of biorelevant media for assessment of a poorly soluble weakly basic drug in the form of liquisolid compacts: In vitro and in vivo study. Drug Delivery, 23: 808-817.
- Khames, A., 2013. Liquisolid technique: A promising alternative to conventional coating for improvement of drug photostability in solid dosage forms. Expert Opin. Drug Delivery, 10: 1335-1343.
- Akpabio E.I., T.O. Uwah, D.E. Effiong and J. Godwin, 2020. Evaluating hydrocolloids of Sida acuta as sustained release matrix for ibuprofen tablet. Global J. Med. Res., 40: 16-21.
- Uwah, T.O.O., E.I. Akpabio, T.C. Jackson, D.E. Effiong and E. Uduk, 2023. Potentials of Sterculia tragacantha Lindl seed husk gum as a release modifier in matrix tablet formulation. GSC Biol. Pharm. Sci., 25: 025-037.
- Karmarkar, A.B., I.D. Gonjari, A.H. Hosmani and P.N. Dhabale, 2010. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Drug Discoveries Ther., 4: 26-32.
- Nokhodchi, A., R. Aliakbar, S. Desai and Y. Javadzadeh, 2010. Liquisolid compacts: The effect of cosolvent and HPMC on theophylline release. Colloids Surf. B: Biointerfaces, 79: 262-269.
- Nnamani, P.O., A.A. Ugwu, E.C. Ibezim, F.C. Kenechukwu and P.A. Akpa et al., 2016. Sustained-release liquisolid compact tablets containing artemether-lumefantrine as alternate-day regimen for malaria treatment to improve patient compliance. Int. J. Nanomed., 11: 6365-6378.
- Aleksić, I., I.G. Ilić, S. Cvijić and J. Parojčić, 2020. An investigation into the influence of process parameters and formulation variables on compaction properties of liquisolid systems. AAPS PharmSciTech, 21.
- Sinha, S., M. Ali, S. Baboota, A. Ahuja, A. Kumar and J. Ali, 2010. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech, 11: 518-527.
- Lam, M., T. Ghafourian and A. Nokhodchi, 2019. Liqui-pellet: The emerging next-generation oral dosage form which stems from liquisolid concept in combination with pelletization technology. AAPS PharmSciTech, 20.
- Azharshekoufeh, L., J. Shokri, M. Barzegar-Jalali and Y. Javadzadeh, 2017. Liquigroud technique: A new concept for enhancing dissolution rate of glibenclamide by combination of liquisolid and co-grinding technologies. Bioimpacts, 7: 5-12.
How to Cite this paper?
APA-7 Style
Effiong,
D.E., Onunkwo,
G.C. (2024). Principles, Applications and Limitations of the Liquisolid System of Drug Delivery: A Review. Trends in Medical Research, 19(1), 178-198. https://doi.org/10.3923/tmr.2024.178.198
ACS Style
Effiong,
D.E.; Onunkwo,
G.C. Principles, Applications and Limitations of the Liquisolid System of Drug Delivery: A Review. Trends Med. Res 2024, 19, 178-198. https://doi.org/10.3923/tmr.2024.178.198
AMA Style
Effiong
DE, Onunkwo
GC. Principles, Applications and Limitations of the Liquisolid System of Drug Delivery: A Review. Trends in Medical Research. 2024; 19(1): 178-198. https://doi.org/10.3923/tmr.2024.178.198
Chicago/Turabian Style
Effiong, Daniel, Ekpa, and Godswill Chukwunweike Onunkwo.
2024. "Principles, Applications and Limitations of the Liquisolid System of Drug Delivery: A Review" Trends in Medical Research 19, no. 1: 178-198. https://doi.org/10.3923/tmr.2024.178.198

This work is licensed under a Creative Commons Attribution 4.0 International License.