Mitophagy in the Pathogenesis of Obesity-Associated Cardiovascular Diseases: New Mechanistic and Therapeutic Insights
-
Kexin Huang
Shanghai Institute of Medicine, Fudan University, Shanghai 200032, China
Jun Ren
National Clinical Research Center for Interventional Medicine, Shanghai 200032, China
| Received 11 Apr, 2024 |
Accepted 18 May, 2024 |
Published 20 May, 2024 |
Mitophagy, a crucial mechanism for maintaining mitochondrial quality, involves the selective removal of damaged and potentially harmful mitochondria through autophagy. Despite its importance, the detailed mechanisms of mitophagy in the heart remain elusive. This review aims to explore the intricacies and therapeutic implications of mitophagy, particularly in the context of obesity, shedding light on its role in preserving cardiac function and cellular homeostasis. This review delves into three main mitophagy pathways: PINK1-PRKN, BNIP3/NIX and FUNDC1-mediated mitophagy. Additionally, this study analyze how obesity affects mitochondrial dynamics and subsequently contributes to cardiac dysfunction. Furthermore, current study discusses potential therapeutic strategies targeting mitophagy to mitigate cardiovascular diseases, hoping to offer new insights into potential therapeutic strategies.
| Copyright © 2024 Huang and Ren. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
Obesity, typically described as the excessive accumulation of fat in the body leading to adverse health effects, is identified by a body mass index (BMI) exceeding 30kg/m2 1. Recent data suggest a persistent and escalating global trend in the prevalence of obesity, mainly influenced by socioeconomic factors and lifestyle modifications. Presently, over 2 billion individuals are classified to be overweight, representing approximately 30% of the global population2,3.
Escalating prevalence of obesity and its association with various comorbidities including type 2 diabetes and cardiovascular diseases (CVDs), render it a global health priority4. A close tie exists between obesity and cardiovascular disorders, notably coronary heart disease, atherosclerosis, atrial fibrillation, hypertension, myocardial infarction (MI) and heart failure (HF)5,6. Conversely, CVD accounts for more than two-thirds of mortalities linked to high BMI, denoting the close tie between heart disease and obesity-related chronic diseases7.
|
Currently, the pathogenesis of CVD in obesity is perceived to be mediated through adipose tissu es, which serve as both paracrine and endocrine organs, producing various adipokines that regulate both metabolic and immunological pathways. For instance, leptin, TNF-α and adiponectin are involved in vascular reactivity modulation, monocyte chemotactic protein 1 and IL-8 may influence inflammation responses, plasminogen activator inhibitors impact the coagulation process. These functions of adipokines are believed to contribute to the development of cardiovascular dysfunction5.
Specific autophagy pathways play a pivotal role in the pathologies of obesity-associated CVDs. Mitophagy, a critical process within mitochondria quality control mechanisms, involves targeted removal of damaged and dysfunctional mitochondria, which may otherwise be cytotoxic8. The term “mitophagy” was initially coined in 2005, following the discovery of Uth1p, a mitochondrial outer membrane protein in yeast that promotes selective autophagic response of mitochondria9. Since then, extensive research has elucidated the mechanisms and functions of mitophagy, revealing its close association with multiple diseases. Mitophagy is regarded as “a cellular self-protective process”, crucial for maintaining mitochondrial metabolic homeostasis and cardiac function during high-fat diet (HFD)-induced cardiomyopathy (obesity cardiomyopathy)8,10.
In cardiac disorders linked to obesity and diabetes mellitus, mitochondrial dysfunction paradoxically triggers aberrant mitophagy. This includes dysfunctional yet beneficial mitophagy, which fails to clear impaired mitochondria, as well as excessive but detrimental mitophagy, resulting in elimination of healthy mitochondria11,12. Furthermore, impairments in autophagy and mitophagy have been linked to a heightened vulnerability of heart to develop various forms of cardiomyopathies spontaneously. Pharmacological or genetic inhibition of autophagy often accelerates disease progression and exacerbates the outcomes of cardiovascular pathologies. These findings underscore the intricate relationship between autophagy, mitophagy and cardiovascular homeostasis8.
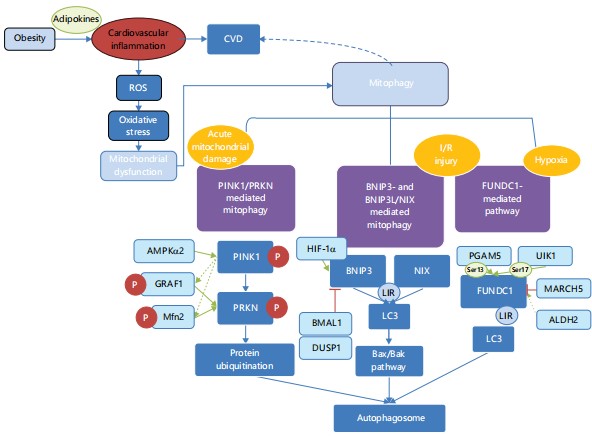
Despite considerable research on the role of mitophagy in obesity-related complications and CVDs, a comprehensive understanding of detailed mechanisms remains elusive. Therefore, this review aims to investigate the intricate relationship between mitophagy and CVDs associated with obesity. It offers a synthesis and critical analysis of the changes in mitophagy observed in cardiac disorders related to obesity, along with their effects and underlying mechanisms (Fig. 1). Additionally, the review aims to provide new insights into potential therapeutic targets in this context.
MITOCHONDRIAL DYSFUNCTION IN OBESITY-RELATED CARDIAC DISORDERS
Mitochondria are essential organelles mainly responsible for regulation of adenosine triphosphate (ATP) production, cell cycle and apoptosis, calcium buffering, generation and elimination of reactive oxygen species (ROS) and antioxidant protection13. Mitochondrial respiratory chain plays a pivotal role in cellular energy generation by coupling electron transfer and ATP synthesis. Amidst this process, the transfer of electrons appears to be concurrent with intracellular ROS production. Respiratory chain complexes I (NADH: Ubiquinone oxidoreductase) and III (ubiquinol: Cytochrome c oxidoreductase) serve as the major sources of ROS. Mitochondrial dysfunction occurs when given mitochondrial genome (mtDNA) and/or nuclear genome (nDNA) are mutated and is featured by compromised mitochondrial biogenesis, disturbed mitochondrial architecture, abnormal mitochondrial DNA (mtDNA) accumulation and excessive ROS accumulation10,13-15. Many of these pathological changes have been noted in obesity and diabetes, suggesting possible underlying etiological mechanisms for the development of cardiovascular complications in obesity.
Obesity is characterized by hypertrophy of adipocytes and infiltration of M1 macrophages, contributing to a state of low-grade chronic inflammation16. Recent research suggests a reciprocal relationship between inflammation and oxidative stress17. Inflammation-related mechanisms can increase the generation of ROS. This oxidative imbalance can lead to mitochondrial dysfunction, a hallmark of many CVDs16. The ROS levels play a crucial role in regulating cardiac activity. While a moderate amount of ROS is essential for normal physiological processes such as cardiac development, cardiomyocyte maturation, cardiac calcium homeostasis, excitation-contraction coupling and modulation of vascular tone18.
Conversely, pathological circumstances characterized by deregulated ROS generation and consequent elevation of ROS levels can precipitate oxidative stress, resulting in mitochondrial dysfunction and apoptosis. Aberrant ROS metabolism and oxidative stress are recognized contributors to multiple cardiac anomalies, including atherosclerosis, hypertension, type 2 diabetes mellitus, cardiac hypertrophy, heart failure (HF), ischemia-reperfusion injury (IRI) and diabetic cardiomyopathy19,20.
Mitophagy mechanisms in cardiovascular physiology: The machinery of mitophagy has not been fully elucidated. In the context of cardiac physiology, three main mitophagy pathways have been recognized: The PINK1-Parkin mediated pathway, the BNIP3-mediated pathway and the FUNDC1-mediated pathway. Each of these pathways and their responses under obesity conditions will be sequentially introduced. Moreover, we cover recent discoveries regarding mitophagy mechanisms, which should enhance and integrate the knowledge network surrounding mitophagy.
PINK1/PRKN-mediated mitophagy: The mitophagy pathway mediated by PTEN induced kinase 1 (PINK1) and parkin RBR E3 ubiquitin protein ligase (PRKN) is considered as the most prevalent mitophagy pathway due to its ubiquity and critical role across various cell types and organisms. The Pink1 gene encodes a 581-amino acid protein that consists of an N-terminal mitochondrial targeting sequence (MTS), a potential transmembrane region, a serine/threonine kinase domain and a highly conserved C-terminal extension. The PINK1 serves as a sensor in the mitophagy pathway. Under normal conditions, the full-length PINK1 is translocated into the inner mitochondrial membrane (IMM) through the maintenance of the mitochondrial membrane potential and is degraded via the ubiquitin-proteasome system21. However, when mitochondria are impaired and therefore depolarized, PINK1 autophosphorylates at Ser228 and accumulates and settles on the mitochondrial membrane, subsequently recruits and phosphorylates PRKN, thereby initiating mitophagy22,23.
The PRKN, an E3 ubiquitin ligase that contains a ubiquitin-like (UBL) domain, acts downstream of PINK1. The UBL domain within PRKN is instrumental in its activation, for it encompasses substrate identification, engagement and attachment to SH3 or ubiquitin interacting motif (UIM) domains and interaction with the proteasome21. The PINK1 phosphorylates PRKN at the Ser65 residue, initiating an intricate signaling cascade through the PRKN-mediated ubiquitination of proteins on outer mitochondrial membrane, ultimately facilitating the sequestration and degradation of compromised mitochondria24,25. Additionally, PINK1 and PRKN engage in mitochondria fusion. Phosphorylation of mitochondrial fusion 2 protein (Mfn2) by PINK1 recruits PRKN to mitochondria, resulting in the ubiquitination and degradation of Mfn2 activity26. The Mfn2 degradation results in an increased mitochondrial fission, which is critical for the packaging and digestion of autophagosome-containing mitochondria. Cardiac knockout of Mfn2 gene compromised PRKN-mediated mitophagy, leading to high ROS production, heart failure and various cardiac dysfunction27.
Multiple factors are involved in PINK1/PRKN-mediated mitophagy in cardiovascular physiology and pathology. The AMP-activated protein kinase (AMPK), functioning as a cellular energy sensor and metabolic regulator, is crucial for cell growth, survival and systemic energy balance. Recent evidence suggests that AMPKα2 interacts with PINK1 by phosphorylating Ser495 and enhances the PINK1/PRKN pathway. In addition, the switch from AMPKα2 to AMPKα1 occurs during the progression of HF, resulting in suppressed AMPKα2 activity and suppressed mitophagy in the chronic phase of HF, denoting therapeutic potential of targeting mitophagy in the management of HF. Another protein closely correlated to PINK1 is GTPase regulator associated with Focal adhesion kinase 1 (GRAF1). Serving as a substrate of PINK1, GRAF1 translocated to the OMM of damaged mitochondria. There, GRAF1 is phosphorylated by PINK1 or PINK1-associated kinases and subsequently directly interacts with PRKN, facilitating Parkin-LC3 interactions to enhance the sequestration and engulfment of damaged mitochondria by autophagosomes28. Thus, disruption of GRAF1 function implicates the pathogenesis of HF, as well as other cardiometabolic or neurological disorders caused by abnormal mitochondrial clearance.
Debate continues regarding the disparity between in vitro and in vivo PINK1/PRKN pathways. Much of the current research delving into the mechanisms relies on in vitro experiments, typically involving the overexpression of PINK1 or PRKN and acute mitochondrial damage22. Although, in vitro experiments have provided comprehensive characterization of PINK1/PRKN-mediated mitophagy, there is still inconclusive evidence from in vivo experiments to support these findings. Current in vivo experiments have focused on animal models using Drosophila, mice, zebrafish and primates. A study conducted in 2006 with Drosophila demonstrated that flies with a Pink1 gene knockout exhibited mitochondrial defects29. Recent studies have indicated that the absence of the Pink1 gene in monkeys leads to lethal outcomes30. However, mt-QC and mt-Keima Pink1 KO mice or mt-Keima Pink1 or prkn KO zebrafish do not display overt defects in basal mitophagy31,32. Therefore, current study hypothesize that discrete basal and stress-evoked mechanisms underlie mitochondrial clearance, leading to a context-dependent process of mitophagy. Consequently, PINK1/PRKN-mediated mitophagy reflects species specificity and is more likely to be activated under severe circumstances, compensating for acute, chemical stress-induced mitochondrial damage. Nevertheless, PINK1/PRKN-mediated mitophagy participates in continuous mitochondrial quality control. Recent research reported that Nuanxinkang (NXK), a Chinese herbal formulation, helps to restore cardiac PINK1/PRNK-mediated mitophagy in MI-induced chronic heart failure (CHF) mice, thereby preventing mitochondrial dysfunction and improving cardiac function33. Further research is needed to validate the extent to which this pathway can function in chronic, cumulative mitochondrial dysfunction.
BNIP3- and BNIP3L/NIX-mediated mitophagy: Apart from PINK1/PRKN-mediated pathway, mitophagy can also be mediated by ubiquitin-independent mitophagy receptors such as Bcl2 interacting protein 3 (BNIP3), BNIP3-like (BNIP3L/NIX) and FUN14 domain-containing 1 (FUNDC1). Being part of the BH3-only protein family, BNIP3 and its homologue NIX are known to regulate cell death and mitophagy. Both BNIP3 and NIX are consisting of a BH3 domain and a C-terminal transmembrane domain. The BH3 domain enables BNIP3 and NIX to interact with anti-apoptotic proteins, thereby promote autophagy and apoptosis. The C-terminal transmembrane domain is particularly significant for mitochondrial OMM localization and proapoptotic function. Moreover, BNIP3 and NIX triggers release of cytochrome c from mitochondria in vitro, leading to loss of mitochondrial membrane potential (ΔΨm)34,35. The BNIP3 and BNIP3L/NIX interplay with LC3 (microtubule-associated proteins 1A/1B light chain 3B) by the LC3 interacting region (LIR) sequence, which activates autophagosome and initiates mitophagy. Both proteins play a pivotal role in the regulation of cell death and mitophagy, contributing to the maintenance of cellular homeostasis and response to stress.
The TBNIP3 functions on both outer and inner mitochondrial membrane (IMM) of mitochondria. In vitro studies have demonstrated that BNIP3 triggers the Bax/Bak pathway by neutralizing anti-apoptotic Bcl-2 proteins, therefore mediating mitochondrial swelling and OMM permeabilization36. Dhingra et al.37 have reported that overexpression of the BNIP3 gene in cardiomyocytes is correlated with diminished mitochondrial membrane potential (ΔΨm) and opening of the mitochondrial permeability transition pore (mPTP), leading to mitochondrial dysfunction and cell death of cardiac myocytes. The insensitivity of BNIP3-induced mitochondrial swelling to cyclosporin A, a mPTP inhibitor, further corroborating the role of BNIP3 in IMM permeabilization. Additionally, Western blot analysis frequently revealed cytochrome c as a doublet in BNIP3-treated samples38. Thus, it is assumed that BNIP3 intervention may elicit unknown redox-driven modifications of cytochrome c. These findings elucidate the molecular mechanisms through which BNIP3 contributes to mitochondria-mediated cell death in the heart.
Recent research has broadened our understanding of BNIP3 and BNIP3L/NIX in cardiovascular physiology, revealing a more comprehensive and extensive network of their functions. Myocardial ischemia manifests in multiple symptoms such as myocardial infarction, angina pectoris and sudden cardiac death. However, blood flow re-establishment after ischemia can provoke additional cardiac harm called myocardial ischemia/reperfusion (I/R) injury. A compromised autophagic flux including I/R injury contributes to various CVDs39. The BNIP3L/NIX-mediated mitophagy plays a critical role in response to cardiac hypoxia. Ischemia/reperfusion (I/R) and oxygen-glucose deprivation/reperfusion (OGD/R) injuries elevated the expression of hypoxia-inducible factor 1 alpha (HIF-1α), subsequently activated BNIP3 signaling cascade and triggered mitophagy40.
Another research also offers similar findings about I/R injury and BNIP3-related mitophagy. Jin et al.41 elaborated that the depletion of dual specificity phosphatase 1 (DUSP1) amplifies the phosphorylation-mediated activation of BNIP3 through JNK, leading to initiating of mitophagy. Chronic intermittent hypoxia (CIH), which may be caused by obstructive sleep apnea syndrome, is prone to induce cardiac hypertrophy. The BNIP3 knockout in rats exacerbated CIH-mediated hypertrophy and cardiac dysfunction, concurrently leading to a notable reduction in autophagy. On the contrary, CIH treatment compensatory upregulated BNIP3 expression, which activates the PI3K/Akt/mTOR pathway, thereby mitigating cardiac hypertrophy42. A recent study has documented the pivotal role of BNIP3L/NIX-mediated mitophagy in glucocorticoid-induced mitochondrial dysfunction, synaptic defects and cellular demise. It has been observed that under high concentrations of glucocorticoids, glucocorticoid receptor (GR) binds to PGC1α promoter, inhibiting PGC1α expression and consequently, a reduction in NIX levels43. This downregulation culminates in decreased mitophagy, highlighting the intricate relationship between glucocorticoids and mitochondrial homeostasis.
The BNIP3 participates in multiple cardiac physiological processes. Circadian rhythm disruption impairs mitophagy and affects cardiac function. The core circadian rhythm gene Bmal1 specifically binds to the E-box element in the promoter region of Bnip3. Deficiency of BMAL1 leads to reduced levels of BNIP3 in cardiomyocytes, resulting in decreased BNIP3-mediated mitophagy and mitochondrial dysfunction and ultimately causing cardiomyocyte disorders44. This study implicates a close association between circadian rhythm factors and mitochondrial activity as well as cardiovascular function. Thus, it provides a potential explanation for the mechanisms by which obesity prompts cardiovascular damages. Several studies have emphasized that disruptions of circadian rhythms are correlated with an elevated vulnerability to a range of health conditions, including CVD, obesity and metabolic disorders45.
Moreover, BNIP3L/NIX-mediated mitophagy emerges as a critical regulatory mechanism essential for the remodeling of the mitochondrial network in cell differentiation. It facilitates the proper reorganization of the mitochondrial network, which is imperative for the functional maturation of cardiac progenitor cells (CPCs). Disruption of this mitophagy process leads to sustained mitochondrial fission and aberrant mitochondrial morphology, resulting in increased susceptibility to cell death and compromised survival of CPCs within MI condition. The study outcome also reveals that the accumulation of mitochondrial DNA (mtDNA) mutations, a hallmark of aging, selectively impairs the differentiation-induced mitophagy program in CPCs15. This finding demonstrates the importance of mitophagy as a pivotal factor in the cellular response to stress and the maintenance of genomic integrity, underscores the indispensable role of BNIP3L/NIX and FUNDC1-mediated mitophagy in regulating the mitochondrial dynamics.
FUNDC1-mediated mitophagy: The FUNDC1, a mitochondria-associated membranes (MAMs)-located protein, is crucial for the maintenance of MAM homeostasis and indispensable in receptor-mediated mitophagy. Similar to BNIP3 and BNIP3/NIX, FUNDC1 binds to LC3 with a LIR sequence, triggering the phosphorylation of three key residues of FUNDC139. This post-transcriptional modification altered the binding affinity of FUNDC1 and LC3 and consequently activates or inhibits mitophagy46. Under physiological conditions, phosphorylation and dephosphorylation at distinct sites ensure low affinity between FUNDC1 and LC339. Moreover, the membrane-associated RING-CH protein 5 (MARCH5) mediates the ubiquitination and subsequent degradation of FUNDC1, inhibiting its activity47. In obese hearts, FUNDC1 interact with the F-box protein FBXL2 to maintain mitochondrial Ca2+ homeostasis and cardiac function48. During pathological conditions such as hypoxia, there are two somewhat independent pathways through which FUNDC1 may govern mitophagy: the kinase-mediated mechanism and the interactional protein-mediated mechanism. The transcriptional and post-transcriptional regulation of FUNDC1 may also play a significant role in the modulation of mitophagy39.
Several studies have reported dephosphorylation of FUNDC1 at Serine 13 by PGAM5 in hypoxia condition, indicating that FUNDC1 is activated and interacted with LC3 motif in hypoxia, mediating selective mitophagy49,50. A recent study reported that BNIP3 and FUNDC1 are selectively activated in mitophagy of cardiac progenitor cells, indicating the significant role of FUNDC1 in CPC homeostasis for the first time15. The fact that CPCs reside in hypoxic environment in vivo further identified the function of FUNDC1-mediated mitophagy in hypoxia. Consequently, the suppression of FUNDC1-mediated mitophagy represents possibility in myocardial I/R damage. In addition, the levels of FUNDC1 phosphorylation at Serine 13 (Ser13) and Tyrosine 18 (Tyr18) suggest a strong correlation with myocardial damage. indicating their potential role as diagnostic and prognostic biomarkers for acute MI.
Platelets are excessively activated during the I/R period. This activation prompts platelets to cluster spontaneously and form microthrombi, which obstruct microvasculature and result in myocardial cell injury or MI. Platelets activation initiates substantial morphological alterations that require significant amount of energy51. Thus, preserving the balance of energy and the structural integrity of mitochondria is vital. On one hand, mitophagy provides a protective effect by eliminating dysfunctional mitochondria, preventing further activation of platelets and the development of thrombosis. On the other hand, a deficiency in mitophagy leads to platelet mitochondrial impairment and a reduction in ATP synthesis, which in turn causes aberrant platelet activation and aggregation52. Zhang et al.53 demonstrated the enhancement of mitophagy within platelets through post-transcriptional modifications of FUNDC1, indicating that FUNDC1-meidated mitophagy is increased in both platelets and cardiomyocytes as a reaction to the challenges posed by ischemia and hypoxia.
Dysfunctional mitochondrial metabolism is a crucial pathological basis of heart failure. Therefore, factors promote mitophagy are considered protective to cardiac function. The Unc-51-like autophagy activating kinase 1 (ULK1), a fundamental serine/threonine kinase in autophagy induction, is specifically involved in the activation of mitophagy, especially under hypoxic conditions54. A previous study demonstrated that Ginsenoside Rg3 triggers the phosphorylation of FUNDC1 at Serine 17 (Ser17) through ULK1-dependent mechanisms, thus activating FUNDC1-LC3 interaction and initiating FUNDC1-mediated mitophagy55. Moreover, Li et al.56 observed that α-LA treatment possibly through a mitochondrial aldehyde dehydrogenase (ALDH2)-dependent mechanism counteracted the reduction of FUNDC1-mediated mitophagy in pressure-overload induced HF. Since ALDH2 is perceived being essential in the etiology of HF whereas α-LA appears to activate FUNDC1 through a NRF1-depedent mechanism, these findings indicate that ALDH2 may possibly regulate FUNDC1 under pathological pressure-overload conditions56.
INTERPLAY BETWEEN MITOPHAGY AND CARDIOVASCULAR COMPLICATIONS OF OBESITY
Obesity-induced mitochondrial dysfunction impairs mitophagy, leading to mitochondrial accumulation and further dysfunction. This vicious cycle promotes cardiovascular complications by affecting cellular metabolism, increasing ROS production and inducing apoptotic cell death.
The Nrf2 transcription factor activated by oxidative stress mediates mitofusin 2 (Mfn2) expression and genes encoding proteasomes. Data has shown that Nrf2 plays a pivotal role in upregulating mitophagy by enhancing the expression of PINK1 and p62 in cardiac dysfunction including MI. Furthermore, Nrf2 functions in mitochondrial biogenesis via influencing the expression of key regulatory factors such as PGC-1α, NResF1, NResF2, TFAM and a range of mitochondrial genes57. A recent study explored the consequences of ALDH2 overexpression (OE) and knockout (KO) in diabetic cardiomyopathy, with a particular emphasis on maintaining mitochondrial integrity58. In vivo and in vitro findings indicated the protective effect of ALDH2 against STZ or high glucose-induced cardiac anomalies. This effect is thought to be mediated through the modulation of mitophagy and maintenance of mitochondrial function, as well as the Akt post-insulin receptor signaling58.
Additionally, obese adipocytes secrete various cytokines and chemokines that further affect mitochondrial function and mitophagy in cardiomyocytes, endothelial cells and vascular smooth muscle cells, contributing to the pathogenesis of CVD. Lipotoxicity is a type of cellular stress that arises from excessive accumulation of lipids, which subsequently leads to mitochondrial dysfunction and insulin resistance within muscle cells. An increase in BNIP3L levels has been observed in the soleus muscle of rats consuming a high-fat (HF) diet, which correlates with the enhancement of mitophagy and the activation the MTOR-RPS6KB pathway, a negative modulator of insulin signaling59.
This finding shed light on the roles of BNIP3L in regulation of mitochondrial dynamics and the association with insulin resistant in tissue, indicating the close relationship between BNIP3L-mediated mitophagy and obesity. More recent research also accords with this conclusion. In type 1 diabetic cardiomyopathy, the depletion of ATP or the surplus of AMP stimulates the activity of AMPK to initiate autophagy, culminating in heart failure60. An overactivation of autophagy under insulin resistance condition can lead to the cardiac cell dysfunction and cytotoxic effects61. However, obesity and high-fat diet-induced insulin resistance condition suppressed the cardiac autophagic flux, which is correlated with elevated p62 levels and reduced LC3II levels61.
THERAPEUTIC INSIGHTS
Targeting mitophagy presents a promising avenue for therapeutic intervention in addressing the cardiovascular complications associated with obesity. Potential strategies include augmenting mitophagy activity, curbing mitochondrial ROS production and reinstating mitochondrial biogenesis. Moreover, addressing obesity-related comorbidities like insulin resistance and dyslipidemia may also contribute to the improvement of mitochondrial function and mitophagy. For example, empagliflozin, a SGLT2 inhibitor, has demonstrated efficacy in reducing ischemia/reperfusion-induced microvascular, endothelial cell damage as well as safeguarding against PD-induced cardiac anomalies by activating FUNDC1-mediated mitophagy via the AMPKα1/ULK1 pathway62,63. These findings open up the prospect of employing empagliflozin in clinical settings to manage acute microvascular dysfunction and other mitophagy-related abnormalities.
Several herbal remedies also function in treating cardiovascular diseases caused by obesity by regulating mitophagy pathways. Baicalin, a natural flavonoid detached from the root of Scutellaria baicalensis, has a protective effect against hypoxia-induced apoptosis in H9c2 cardiomyocytes. It is speculated that baicalin activated the Nrf2/HO-1 and HIF1α/BNIP3 signaling cascades64. Baicalin is proved to ameliorate cell viability loss and the apoptotic effects under hypoxic conditions, providing a valuable insight into myocardial ischemia. Danqi Pill (DQP), constituted by dry roots of Salvia miltiorrhiza Bunge and Panax notoginseng, restored mitophagy in HF rats in vivo. Further in vitro experiments elucidate that DQP increases the interaction between FUNDC1 and ULK1 or PGAM5, indicating the role of DQP in protecting against HF by promoting mitophagy mediated by FUNDC165.
CONCLUSION
The intricate relationship between mitophagy and obesity-associated cardiovascular diseases is increasingly acknowledged as a critical determinant in the initiation and progression of these conditions. Mitophagy, a selective form of autophagy targeting damaged mitochondria, serves a multifaceted role in preserving cardiac function and influencing disease advancement under diverse metabolic challenges. This review has underscored the diverse modalities of mitophagy, particularly emphasizing the PINK1-PRKN, BNIP3/NIX and FUNDC1 pathways and elucidate how their dysregulation contributes to mitochondrial dysfunction and subsequent development of CVD. Furthermore, we have highlighted the potential therapeutic implications of modulating mitophagy in the context of obesity-related heart diseases. Emerging evidence from both pharmacological and herbal interventions suggests that enhancing mitophagy holds promise in improving mitochondrial quality control and cardiac function, offering hopeful avenues for treating the cardiovascular complications of obesity. However, future research endeavors are essential to unravel the precise molecular mechanisms governing mitophagy in obesity-related CVDs and identify novel therapeutic targets that can be translated into clinical practice for the benefit of patients.
SIGNIFICANCE STATEMENT
This review focuses on mitophagy machinery in obesity-associated cardiovascular diseases and probes into the intricate interplay among obesity, mitochondrial dynamics and cardiac function. The process of mitophagy in cardiac homeostasis is of great significance although many aspects related to mitophagy in cardiovascular medicine remains unclear at this time. Therefore, this review aims to fill the gap by offering a synthesis and critical analysis of mitophagy pathways triggered in obesity-assocciated cardiovascular diseases, along with the underlying mechanisms. Moreover, this review hopes to spark novel insights into potential therapeutic approaches for cardiovascular diseases.
REFERENCES
- Piché, M.E., A. Tchernof and J.P. Després, 2020. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ. Res., 126: 1477-1500.
- Caballero, B., 2019. Humans against obesity: Who will win? Adv. Nutr., 10: S4-S9.
- Ren, J., N.N. Wu, S. Wang, J.R. Sowers and Y. Zhang, 2021. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev., 101: 1745-1807.
- Perdomo, C.M., R.V. Cohen, P. Sumithran, K. Clément and G. Frühbeck, 2023. Contemporary medical, device, and surgical therapies for obesity in adults. Lancet, 401: 1116-1130.
- Mandviwala, T., U. Khalid and A. Deswal, 2016. Obesity and cardiovascular disease: A risk factor or a risk marker? Curr. Atheroscler. Rep., 18.
- Blüher, M., 2019. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol., 15: 288-298.
- The GBD 2015 Obesity Collaborators, 2017. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med., 377: 13-27.
- Pedro, J.M.B.S., G. Kroemer and L. Galluzzi, 2017. Autophagy and mitophagy in cardiovascular disease. Circ. Res., 120: 1812-1824.
- Lemasters, J.J., 2005. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res., 8: 3-5.
- Tang, S., D. Hao, W. Ma, L. Liu and J. Gao et al., 2024. Dysfunctional mitochondria clearance in situ: Mitophagy in obesity and diabetes-associated cardiometabolic diseases. Diabetes Metab. J., 2024.
- Hombrebueno, J.R., L. Cairns, L.R. Dutton, T.J. Lyons and D.P. Brazil et al., 2019. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight, 4.
- Shao, D., S.C. Kolwicz, P. Wang, N.D. Roe and O. Villet et al., 2020. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating parkin-mediated mitophagy. Circulation, 142: 983-997.
- Das, M., C. Sauceda and N.J.G. Webster, 2021. Mitochondrial dysfunction in obesity and reproduction. Endocrinology, 162.
- Liu, Y., Y. Huang, C. Xu, P. An and Y. Luo et al., 2022. Mitochondrial dysfunction and therapeutic perspectives in cardiovascular diseases. Int. J. Mol. Sci., 23.
- Lampert, M.A., A.M. Orogo, R.H. Najor, B.C. Hammerling and L.J. Leon et al., 2019. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy, 15: 1182-1198.
- de Mello, A.H., A.B. Costa, J.D.G. Engel and G.T. Rezin, 2018. Mitochondrial dysfunction in obesity. Life Sci., 192: 26-32.
- Bondia-Pons, I., L. Ryan and J.A. Martinez, 2012. Oxidative stress and inflammation interactions in human obesity. J. Physiol. Biochem., 68: 701-711.
- Burgoyne, J.R., H. Mongue-Din, P. Eaton and A.M. Shah, 2012. Redox signaling in cardiac physiology and pathology. Circ. Res., 111: 1091-1106.
- Forman, H.J. and H. Zhang, 2021. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discovery, 20: 689-709.
- Peoples, J.N., A. Saraf, N. Ghazal, T.T. Pham and J.Q. Kwong, 2019. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med., 51: 1-13.
- Liang, Y., G. Zhong, M. Ren, T. Sun and Y. Li et al., 2023. The role of ubiquitin-proteasome system and mitophagy in the pathogenesis of Parkinson's disease. Neuromol. Med., 25: 471-488.
- Han, R., Y. Liu, S. Li, X.J. Li and W. Yang, 2023. PINK1-PRKN mediated mitophagy: Differences between in vitro and in vivo models. Autophagy, 19: 1396-1405.
- Gan, Z.Y., S. Callegari, S.A. Cobbold, T.R. Cotton and M.J. Mlodzianoski et al., 2022. Activation mechanism of PINK1. Nature, 602: 328-335.
- Ajoolabady, A., M. Chiong, S. Lavandero, D.J. Klionsky and J. Ren, 2022. Mitophagy in cardiovascular diseases: Molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med., 28: 836-849.
- Kakade, P., H. Ojha, O.G. Raimi, A. Shaw and A.D. Waddell et al., 2022. Mapping of a N-terminal α-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol., 12.
- Saito, T. and J. Sadoshima, 2015. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res., 116: 1477-1490.
- Chen, Y. and G.W. Dorn, 2013. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science, 340: 471-475.
- Zhu, Q., M.E. Combs, J. Liu, X. Bai and W.B. Wang et al., 2023. GRAF1 integrates PINK1-Parkin signaling and actin dynamics to mediate cardiac mitochondrial homeostasis. Nat. Commun., 14.
- Clark, I.E., M.W. Dodson, C. Jiang, J.H. Cao and J.R. Huh et al., 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature, 441: 1162-1166.
- Yang, W., Y. Liu, Z. Tu, C. Xiao and S. Yan et al., 2019. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res., 29: 334-336.
- Wrighton, P.J., A. Shwartz, J.M. Heo, E.D. Quenzer, K.A. LaBella, J.W. Harper and W. Goessling, 2021. Quantitative intravital imaging in zebrafish reveals in vivo dynamics of physiological-stress-induced mitophagy. J. Cell Sci., 134.
- McWilliams, T.G., A.R. Prescott, L. Montava-Garriga, G. Ball and F. Singh et al., 2018. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab., 27: 439-449.E5.
- Guan, Z., J. Chen, L. Wang, M. Hao and X. Dong et al., 2023. Nuanxinkang prevents the development of myocardial infarction-induced chronic heart failure by promoting PINK1/Parkin-mediated mitophagy. Phytomedicine, 108.
- Zhang, J. and P.A. Ney, 2009. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ., 16: 939-946.
- Gao, A., J. Jiang, F. Xie and L. Chen, 2020. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta, 506: 72-83.
- O'Neill, K.L., K. Huang, J. Zhang, Y. Chen and X. Luo, 2016. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev., 30: 973-988.
- Dhingra, A., R. Jayas, P. Afshar, M. Guberman and G. Maddaford et al., 2017. Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radical Biol. Med., 112: 411-422.
- Quinsay, M.N., Y. Lee, S. Rikka, M.R. Sayen, J.D. Molkentin, R.A. Gottlieb and Å.B. Gustafsson, 2010. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J. Mol. Cell. Cardiol., 48: 1146-1156.
- Li, G., J. Li, R. Shao, J. Zhao and M. Chen, 2021. FUNDC1: A promising mitophagy regulator at the mitochondria-associated membrane for cardiovascular diseases. Front. Cell Dev. Biol., 9.
- Zhang, Y., D. Liu, H. Hu, P. Zhang, R. Xie and W. Cui, 2019. HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother., 120.
- Jin, Q., R. Li, N. Hu, T. Xin and P. Zhu et al., 2018. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol., 14: 576-587.
- Chi, R., C. Chai, G. Liu, H. Cao and B. Yang, 2023. Chronic intermittent hypoxia-induced BNIP3 expression mitigates contractile dysfunction and myocardial injury in animal and cell model via modulating autophagy. Hum. Cell, 36: 631-642.
- Choi, G.E., H.J. Lee, C.W. Chae, J.H. Cho and Y.H. Jung et al., 2021. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat. Commun., 12.
- Li, E., X. Li, J. Huang, C. Xu and Q. Liang et al., 2020. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell, 11: 661-679.
- Miro, C., A. Docimo, L. Barrea, L. Verde and S. Cernea et al., 2023. “Time” for obesity-related cancer: The role of the circadian rhythm in cancer pathogenesis and treatment. Semin. Cancer Biol., 91: 99-109.
- Wang, J., P. Zhu, R. Li, J. Ren and H. Zhou, 2020. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol., 30.
- Chen, Z., S. Siraj, L. Liu and Q. Chen, 2017. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy, 13: 1244-1245.
- Ren, J., M. Sun, H. Zhou, A. Ajoolabady and Y. Zhou et al., 2020. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-depen-dent manner in obesity. Sci. Adv., 6.
- Liu, L., D. Feng, G. Chen, M. Chen and Q. Zheng et al., 2012. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell. Biol., 14: 177-185.
- Chen, G., Z. Han, D. Feng, Y. Chen and L. Chen et al., 2014. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell, 54: 362-377.
- Zhang, W., S. Siraj, R. Zhang and Q. Chen, 2017. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy, 13: 1080-1081.
- Mao, Y., J. Ren and L. Yang, 2022. FUN14 domain containing 1 (FUNDC1): A promising mitophagy receptor regulating mitochondrial homeostasis in cardiovascular diseases. Front. Pharmacol., 13.
- Zhang, W., H. Ren, C. Xu, C. Zhu and H. Wu et al., 2016. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. elife, 5.
- Deng, R., H.L. Zhang, J.H. Huang, R.Z. Cai and Y. Wang et al., 2021. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy, 17: 3011-3029.
- Wang, X., G. Ling, Y. Wei, W. Li and Y. Zhang et al., 2023. Activation of ULK1 to trigger FUNDC1-mediated mitophagy in heart failure: Effect of Ginsenoside Rg3 intervention. Phytomedicine, 120.
- Li, W., L. Yin, X. Sun, J. Wu and Z. Dong et al., 2020. Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis., 11.
- Chen, Q.M., 2022. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radical Biol. Med., 179: 133-143.
- Zhang, Y., R. Zou, M. Abudureyimu, Q. Liu and J. Ma et al., 2023. Mitochondrial aldehyde dehydrogenase rescues against diabetic cardiomyopathy through GSK3β-mediated preservation of mitochondrial integrity and parkin-mediated mitophagy. J. Mol. Cell Biol.
- da Silva Rosa, S.C., M.D. Martens, J.T. Field, L. Nguyen and S.M. Kereliuk et al., 2021. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy, 17: 2257-2272.
- Kanamori, H., G. Takemura, K. Goto, A. Tsujimoto and A. Mikami ety al., 2015. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy, 11: 1146-1160.
- Sadeghi, A., M. Niknam, M.A. Momeni-Moghaddam, M. Shabani and H. Aria et al., 2023. Crosstalk between autophagy and insulin resistance: Evidence from different tissues. Eur. J. Med. Res., 28.
- Cai, C., Z. Guo, X. Chang, Z. Li and F. Wu et al., 2022. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol., 52.
- Yu, W., L. Wang, W.Y. Ren, H.X. Xu and N.N. Wu et al., 2024. SGLT2 inhibitor empagliflozin alleviates cardiac remodeling and contractile anomalies in a FUNDC1-dependent manner in experimental Parkinson’s disease. Acta Pharmacol. Sin., 45: 87-97.
- Yu, H., B. Chen and Q. Ren, 2019. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1α/BNIP3 pathway. Artif. Cells Nanomed. Biotechnol., 47: 3657-3663.
- Wang, X., Y. Jiang, Y. Zhang, Q. Sun and G. Ling et al., 2022. The roles of the mitophagy inducer Danqi pill in heart failure: A new therapeutic target to preserve energy metabolism. Phytomedicine, 99.
How to Cite this paper?
APA-7 Style
Huang,
K., Ren,
J. (2024). Mitophagy in the Pathogenesis of Obesity-Associated Cardiovascular Diseases: New Mechanistic and Therapeutic Insights. Trends in Medical Research, 19(1), 112-123. https://doi.org/10.3923/tmr.2024.112.123
ACS Style
Huang,
K.; Ren,
J. Mitophagy in the Pathogenesis of Obesity-Associated Cardiovascular Diseases: New Mechanistic and Therapeutic Insights. Trends Med. Res 2024, 19, 112-123. https://doi.org/10.3923/tmr.2024.112.123
AMA Style
Huang
K, Ren
J. Mitophagy in the Pathogenesis of Obesity-Associated Cardiovascular Diseases: New Mechanistic and Therapeutic Insights. Trends in Medical Research. 2024; 19(1): 112-123. https://doi.org/10.3923/tmr.2024.112.123
Chicago/Turabian Style
Huang, Kexin, and Jun Ren.
2024. "Mitophagy in the Pathogenesis of Obesity-Associated Cardiovascular Diseases: New Mechanistic and Therapeutic Insights" Trends in Medical Research 19, no. 1: 112-123. https://doi.org/10.3923/tmr.2024.112.123

This work is licensed under a Creative Commons Attribution 4.0 International License.